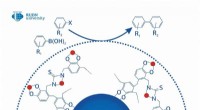
Equipe de pesquisa desenvolve método universal e preciso para calcular como as proteínas interagem com medicamentos
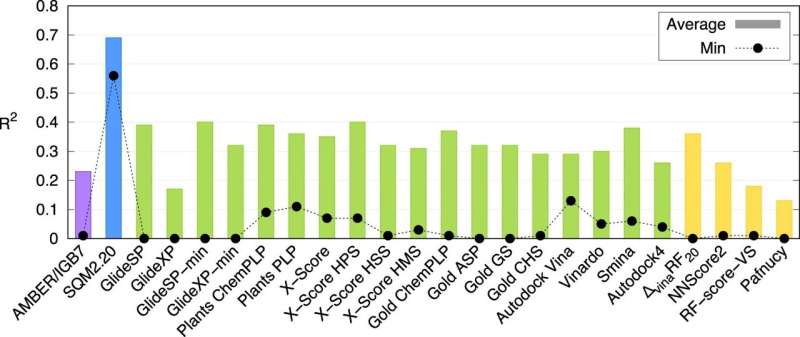
Correlações médias (colunas) e mínimas (círculos pretos) (R2) sobre o conjunto de dados PL-REX. Crédito:Nature Communications (2024). DOI:10.1038/s41467-024-45431-8 Uma equipe de pesquisa do Instituto de Química Orgânica e Bioquímica da Academia Tcheca de Ciências / IOCB Praga desenvolveu um novo método computacional que pode descrever com precisão como as proteínas interagem com moléculas de possíveis medicamentos e pode fazê-lo em apenas algumas dezenas de minutos. Esta nova função de pontuação da mecânica quântica pode, assim, acelerar significativamente a busca por novos medicamentos. A pesquisa foi publicada na revista Nature Communications .
O estudo demonstra que este é o primeiro método desse tipo universalmente aplicável. Os especialistas computacionais do IOCB Prague testaram-no em 10 proteínas de diferentes níveis de complexidade estrutural, cada uma ligando uma grande variedade de pequenas moléculas (geralmente chamadas de ligantes). Eles então compararam seus resultados não apenas com os de outros métodos correspondentes, mas também com os resultados de experimentos de laboratório, e ambas as comparações foram muito favoráveis.
"É claro que não somos os únicos a trabalhar nisso. Existem vários métodos desse tipo. Geralmente, porém, sua velocidade é compensada pela baixa precisão, enquanto cálculos mais precisos podem levar vários dias. Nossos métodos são únicos porque podem processar informações sobre grandes sistemas moleculares em dezenas de minutos, mantendo os benefícios de cálculos de mecânica quântica muito mais exigentes", explica Jan Řezáč, autor correspondente do artigo do grupo de Interações Não-Covalentes liderado pelo Prof. Pavel Hobza. Jan Řezáč do IOCB Praga sobre SQM2.20. Crédito:IOCB Praga Especialistas desse grupo estudam interações intermoleculares há muito tempo. Nesta pesquisa eles se concentram principalmente em biomoléculas, e os resultados de seu trabalho estão diretamente relacionados ao projeto de medicamentos auxiliado por computador. A razão é que quando os cientistas trabalham para desenvolver um novo medicamento, muitas vezes procuram moléculas que se liguem fortemente a uma proteína específica.
Identificá-los, no entanto, é como encontrar agulhas num palheiro, pois é necessário testar um grande número de moléculas para distinguir aquelas que se mostram promissoras. Isto retarda consideravelmente a descoberta de substâncias medicinais e torna-a mais cara. Ao prever a força da ligação proteína-ligante e, assim, selecionar as moléculas que melhor satisfazem um conjunto definido de critérios, os químicos computacionais poupam o trabalho dos experimentadores, o que, por sua vez, acelera significativamente a descoberta de medicamentos.
Mais informações: Adam Pecina et al, SQM2.20:A função de pontuação mecânica quântica semiempírica produz previsões de afinidade de ligação proteína-ligante com qualidade DFT em minutos, Nature Communications (2024). DOI:10.1038/s41467-024-45431-8 Informações do diário: Comunicações da Natureza
Fornecido pelo Instituto de Química Orgânica e Bioquímica do CAS