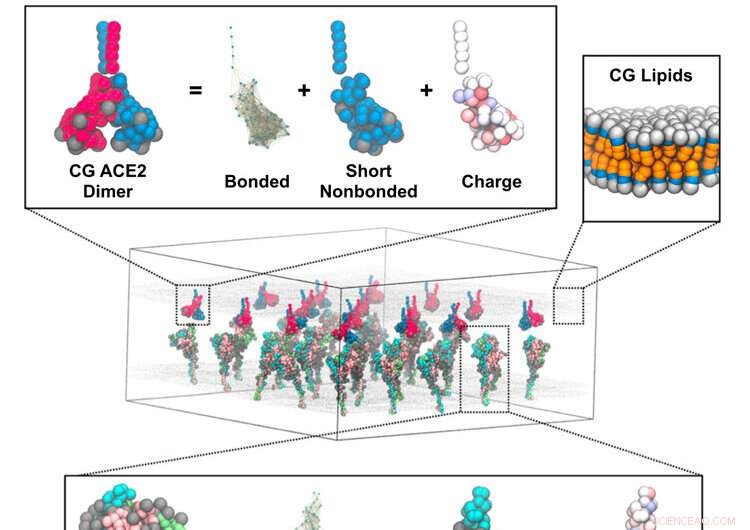
Simulações de supercomputadores revelam os detalhes da fusão do coronavírus
O mecanismo pelo qual o coronavírus se funde às células hospedeiras foi sugerido por meio de simulações de pesquisadores da Universidade de Chicago usando o supercomputador Frontera no TACC. Representação representativa de uma simulação de granulação de curso (CG) de trímeros de pico na membrana interagindo com uma membrana adjacente com dímeros de ACE2. As inserções descrevem os componentes do modelo CG para o trímero de pico (inferior), dímero ACE2 (superior esquerdo) e membrana lipídica (superior direito). Crédito:Pak, A.J., Yu, A., Ke, Z. et al.
O mistério de como exatamente o vírus SARS-CoV-2 infecta as células pulmonares humanas permanece em grande parte oculto para os cientistas experimentais. Agora, no entanto, os detalhes diabólicos do mecanismo pelo qual o coronavírus se funde às células hospedeiras foram sugeridos por meio de simulações de pesquisadores da Universidade de Chicago usando o supercomputador Frontera no Texas Advanced Computing Center (TACC). Os modelos de computador mostram o comportamento cooperativo das proteínas receptoras da célula hospedeira que leva à sua própria infecção. O trabalho pode ser aplicado para ajudar a entender o aumento da virulência de variantes de coronavírus, como delta, omicron e muito mais.
"Descobrimos que a proteína spike interage com dois receptores ACE2 de forma muito cooperativa", disse Gregory Voth, um distinto professor de química da Universidade de Chicago. "Esta é uma visão biofísica fundamental."
Voth é autor sênior do estudo que modelou as interações do coronavírus e das células receptoras com simulações de computador publicadas na revista Nature Communications em fevereiro de 2022.
Como uma bola de futebol com picos, as proteínas dos picos adornam a superfície do coronavírus. Os picos procuram e se fundem com os receptores da proteína da enzima conversora de angiotensina 2 (ACE2) nas células pulmonares humanas. A proteína spike é composta por duas partes principais. O domínio S1 contém o domínio de ligação ao receptor que reconhece as proteínas ACE2. E o domínio S2 contém a maquinaria de fusão, que é protegida e coberta como uma casca pelo domínio S1.
As simulações revelam como uma proteína do receptor ACE2 mantém o pico de coronavírus e o enfraquece enquanto a outra começa a separá-lo. O domínio S1 então se desfaz e expõe a maquinaria de fusão. Este "um-dois" soco prepara o vírus para fusão e entrada nas células hospedeiras do pulmão humano.
"Parece que variantes como delta e omicron podem acentuar ainda mais esse comportamento - é um passo fundamental. Em última análise, futuros anticorpos e, possivelmente, produtos farmacêuticos moleculares devem ser capazes de interferir nesse processo", disse Voth. Filme da dissociação do domínio da proteína S1 do pico de coronavírus induzida pela ligação multivalente de ACE2 durante simulações de dinâmica molecular de granulação grossa. Dois protômeros dentro de dímeros de ACE2 representados como contas vermelhas e azuis. Três protômeros S1 dentro do trímero de pico representados como contas ciano, rosa e verde. Glicanos e protômeros S2 representados por esferas cinza e prata, respectivamente. Lipídios não mostrados. Crédito:Pak, A.J., Yu, A., Ke, Z. et al. Voth e seus colegas desenvolveram o que chamam de "modelos de granulação grossa de baixo para cima" que obtiveram dados de tomografia crio-eletrônica do laboratório do co-autor do estudo John Briggs, do Instituto Max Planck de Bioquímica. Eles o combinaram com simulações de dinâmica molecular atomística. Os dados gerados alimentaram um arcabouço teórico que desenvolveu os modelos de granulação grossa.
"Os modelos de granulação grossa são até 1.000 vezes mais rápidos do que as simulações de dinâmica molecular atomística direta, mas mantêm as características físicas essenciais", disse Voth. Este método proporciona uma enorme economia de tempo e dinheiro nos cálculos.
A equipe científica foi premiada com recursos e serviços de supercomputadores pelo COVID-19 HPC Consortium, um esforço público-privado em apoio à pesquisa COVID-19. Através do consórcio, eles usaram o sistema Frontera financiado pela National Science Foundation no TACC; o cluster de computadores Witherspoon na IBM Research; e recursos do Oak Ridge Leadership Computing Facility no Oak Ridge National Laboratory.
"Nós computamos dados de dinâmica molecular de todos os átomos no Frontera e usamos ferramentas de análise disponíveis no TACC - ambos foram muito valiosos", disse Voth.
A equipe de Voth apresentou seu artigo antes que as variantes delta e omicron fossem conhecidas e, portanto, não previram as mutações. Mas eles voltaram e revisaram os modelos para investigar as variantes.
“Delta tem algo como uma abertura no pico que acontece mais prontamente do que em mutações anteriores do coronavírus”, disse Voth. “Foi emocionante do ponto de vista científico ver comportamentos que não haviam sido vistos antes”.
Voth se referiu a dados de laboratório de microscopia crioeletrônica mostrando a estrutura de uma proteína spike solúvel com dois receptores ACE2 ligados a ela. Mas ele distinguiu esse exemplo cristalizado do que investigou usando simulações no ambiente mais realista de muitas proteínas interagindo umas com as outras em folhas de membrana.
"Os supercomputadores, se bem usados e baseados em boa física, podem fornecer uma maneira totalmente nova de olhar para esses processos. Por meio de simulação por computador, pode-se estudar coisas que não podem ser feitas atualmente com experimentos. Simulação e experimentos funcionam muito bem juntos, de mãos dadas", disse Voth. + Explorar mais
Primeiro modelo completo de coronavírus mostra cooperação