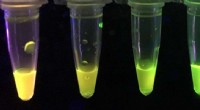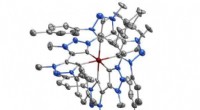
Pesquisadores do laboratório de Jianmin Cui analisaram os mecanismos por trás da função e disfunção de um grupo de proteínas, bem como suas interações com uma droga antiepiléptica, para desenvolver uma nova estratégia potencial para tratar a epilepsia. Crédito:Laboratório Cui
A epilepsia é um distúrbio neurológico que surge da atividade elétrica anormal no cérebro, levando a convulsões. Esses eventos de convulsão podem ter uma variedade de causas, incluindo variantes genéticas em uma família de proteínas que regulam os íons de potássio no cérebro. Pesquisadores da Universidade de Washington em St. Louis lideraram uma equipe internacional para examinar de perto os mecanismos por trás da função e disfunção dessas proteínas, bem como suas interações com uma droga antiepiléptica, para desenvolver uma nova estratégia potencial para tratar a epilepsia.
Jianmin Cui, professor de engenharia biomédica na Escola de Engenharia McKelvey, e Nien-Du Yang, estudante de doutorado em engenharia biomédica que realiza pesquisas no laboratório de Cui, se uniram a Harley Kurata, professor associado de farmacologia da Universidade de Alberta, e investigaram o mecanismo de trabalho de dois canais de íons de potássio, KCNQ2 e KCNQ3. Suas descobertas revelam um mecanismo conservado para a ativação do canal KCNQ que é um alvo tanto de mutações ligadas à epilepsia quanto de um composto de pequenas moléculas.
O trabalho foi publicado em 20 de julho na revista
Science Advances .
A família de canais de potássio KCNQ tem múltiplas funções, desde regular o batimento cardíaco (por KCNQ1) até controlar a excitabilidade dos neurônios (por KCNQ2-5). Esses canais são ativados por voltagem de modo que detectam mudanças de voltagem através da membrana celular e abrem e fecham em resposta. A comunicação entre a detecção de voltagem e a abertura do poro do canal é conhecida como acoplamento eletromecânico, um processo que envolve mudanças conformacionais da proteína durante a ativação dependente de voltagem.
A equipe de Cui mostrou anteriormente que KCNQ1, a isoforma cardíaca KCNQ, apresenta um processo de dois estágios no acoplamento eletromecânico que leva a dois estados abertos de canais distintos, o intermediário-aberto e ativado-aberto. A regulação dos dois estados abertos está subjacente às modulações específicas de tecido do KCNQ1, patogênese da doença e farmacologia. KCNQ2 e KCNQ3 são altamente expressos no sistema nervoso central e são os principais contribuintes para a corrente M, uma corrente crítica de potássio que modula a excitabilidade neuronal. Portanto, a função da corrente M prejudicada por mutações congênitas em KCNQ2 e KCNQ3 é comumente associada à epilepsia de início precoce e pediátrica.
"Embora os canais KCNQ sejam altamente semelhantes em suas sequências e estruturas, não está claro se as isoformas neuronais KCNQ também compartilham o mesmo mecanismo de acoplamento eletromecânico ou dois estados abertos", disse Yang, o primeiro autor do artigo. "Este trabalho revela as principais semelhanças e diferenças entre esses canais que podem ter implicações importantes para sua função em cardiomiócitos ou neurônios".
A equipe usou uma variedade de métodos para estudar o mecanismo de acoplamento eletromecânico nesses canais de potássio, incluindo a criação de mutações genéticas específicas nos canais, eletrofisiologia e medições ópticas de fluorescência.
"Elucidar o mecanismo molecular para o acoplamento eletromecânico é um passo importante para a compreensão do gating dependente da voltagem dos canais de potássio", disse Cui. "Nós fornecemos evidências funcionais de que os canais neuronais KCNQ2 e KCNQ3 são diferentes de KCNQ1 em que apresentam um único estado aberto ativado, mas com um mecanismo de acoplamento eletromecânico conservado específico para o estado aberto ativado".
Esses canais são os principais alvos para tratamentos para epilepsia, descobriram os pesquisadores. A equipe também identificou um conjunto de mutações em KCNQ2 e KCNQ3 associadas à encefalopatia epiléptica infantil precoce, uma forma grave de epilepsia infantil, que interrompe especificamente o acoplamento eletromecânico dos canais. Os pesquisadores aproveitaram um protótipo de droga antiepiléptica retigabina dado seu mecanismo de ação nos canais neuronais KCNQ e demonstraram que o acoplamento eletromecânico pode ser diretamente aprimorado para resgatar a função desses mutantes doentes. Seus estudos sugerem que o mecanismo de acoplamento eletromecânico nos canais KCNQ pode ser um alvo eficaz, apresentando uma nova estratégia farmacológica para o desenvolvimento de terapias mais eficazes para o tratamento da epilepsia.
+ Explorar mais Disfunção do canal de potássio na epilepsia genética