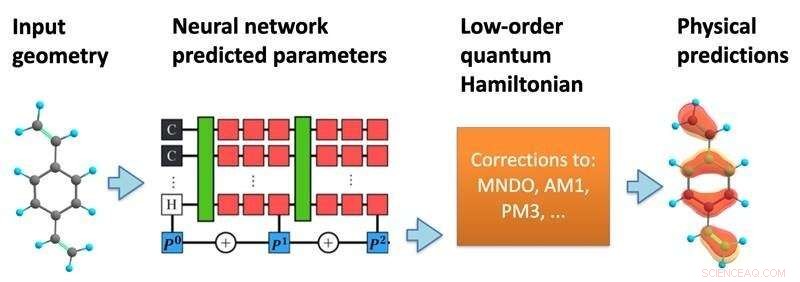
A estrutura do modelo. Uma rede neural processa uma geometria molecular para prever um Hamiltoniano quântico semi-empírico, que é então resolvido de forma auto-consistente para prever uma variedade de propriedades químicas. Crédito:Kipton Barros, Laboratório Nacional de Los Alamos.
Em um novo estudo, publicado em
Proceedings of the National Academy of Sciences , pesquisadores do Laboratório Nacional de Los Alamos propuseram incorporar mais da matemática da mecânica quântica na estrutura das previsões de aprendizado de máquina. Usando as posições específicas dos átomos dentro de uma molécula, o modelo de aprendizado de máquina prevê uma matriz hamiltoniana efetiva, que descreve os vários estados eletrônicos possíveis junto com suas energias associadas.
Em comparação com as simulações tradicionais de química quântica, a abordagem baseada em aprendizado de máquina faz previsões com um custo computacional muito reduzido. Ele permite previsões quantitativamente precisas sobre as propriedades do material, permite uma visão interpretável da natureza da ligação química entre os átomos e pode ser usado para prever outros fenômenos complexos, como como o sistema responderá a perturbações, como interações luz-matéria. O método também fornece precisão muito melhor em relação aos modelos tradicionais de aprendizado de máquina e demonstra sucesso na transferibilidade, ou seja, a capacidade do modelo de fazer previsões que vão muito além dos dados que formaram a base de seu treinamento.
As equações da mecânica quântica fornecem um roteiro para prever as propriedades dos produtos químicos a partir de teorias científicas básicas. No entanto, essas equações podem rapidamente se tornar muito caras em termos de tempo e energia do computador quando usadas para prever o comportamento em grandes sistemas. O aprendizado de máquina oferece uma abordagem promissora para acelerar essas simulações em larga escala. O uso de aprendizado de máquina para prever propriedades químicas tem potencial para grandes avanços tecnológicos, com aplicações desde energia mais limpa até design de medicamentos farmacêuticos mais rápido. Esta é uma área de pesquisa altamente ativa, mas a maioria das abordagens existentes usa abordagens simples e heurísticas para o design dos modelos de aprendizado de máquina.
Em seu estudo, os pesquisadores mostraram que os modelos de aprendizado de máquina podem imitar a estrutura básica das leis fundamentais da natureza. Essas leis podem ser muito difíceis de simular diretamente. A abordagem de aprendizado de máquina permite previsões fáceis de calcular e precisas em uma ampla variedade de sistemas químicos.
O modelo de aprendizado de máquina aprimorado pode prever com rapidez e precisão uma ampla gama de propriedades das moléculas. Essas abordagens pontuam muito bem em importantes benchmarks em química computacional e mostram como os métodos de aprendizado profundo podem continuar a melhorar incorporando mais dados de experimentos. O modelo também pode ser bem-sucedido em tarefas desafiadoras, como prever a dinâmica do estado excitado – como os sistemas se comportam com níveis de energia elevados. Esta ferramenta é uma capacidade inovadora para a química quântica. Isso permitirá que os pesquisadores entendam melhor a reatividade e os estados excitados de novas moléculas.
+ Explorar mais Os computadores se destacam nas aulas de química