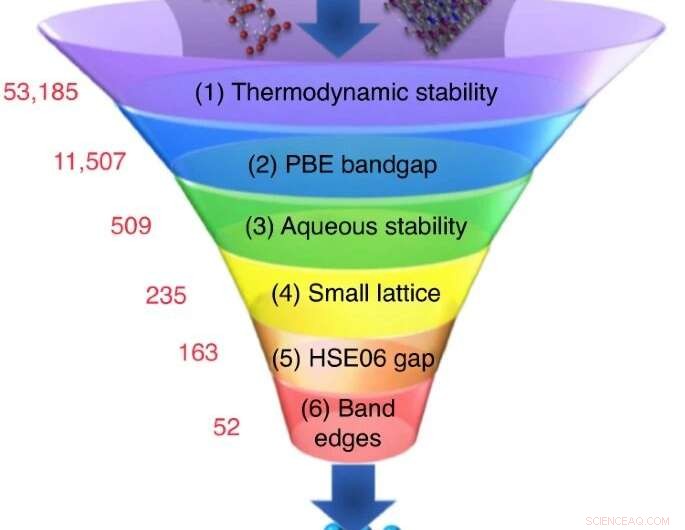
Um grande número de materiais candidatos são escolhidos a partir de bancos de dados experimentais ou computacionais, e uma sequência de cálculos de triagem reduz seu número a um pequeno conjunto de candidatos com as propriedades mais promissoras. Crédito:Nicola Marzari
Nicola Marzari, chefe do laboratório de Teoria e Simulação de Materiais da EFPL e diretor da NCCR MARVEL, acaba de publicar uma revisão dos métodos de estrutura eletrônica como parte de uma edição especial do Insight on Computational Materials Design, publicado por Materiais da Natureza . O artigo, escrito com Andrea Ferretti do CNR – Instituto Nanoscienze e Chris Wolverton da Northwestern University, fornece uma visão geral desses métodos, discute sua aplicação para a previsão de propriedades de materiais, e examina diferentes estratégias usadas para atingir os objetivos mais amplos de design e descoberta de materiais. Olhando para a frente, os autores consideram os desafios emergentes na precisão preditiva dos cálculos, e em abordar a complexidade da vida real de materiais e dispositivos. Eles também enfatizam a importância das infraestruturas computacionais que suportam tais pesquisas, e como o planejamento para financiar esses e os modelos de carreiras de apoio está apenas começando a surgir.
Nos últimos 20 anos, simulações de primeiros princípios tornaram-se poderosas, ferramentas amplamente utilizadas em muitos, diversos campos da ciência e da engenharia. Da nanotecnologia à ciência planetária, da metalurgia aos materiais quânticos, eles aceleraram a identificação, caracterização, e otimização de materiais enormemente. Eles levaram a previsões surpreendentes - do transporte térmico ultrarrápido à supercondutividade mediada por elétron-fônon em hidretos e ao surgimento de bandas planas no grafeno de dupla camada torcida - que inspiraram experimentos notáveis.
O impulso atual para complementar experimentos com simulações; contínuo, rápido crescimento na capacidade de processamento do computador; a capacidade de aprendizado de máquina e inteligência artificial de acelerar a descoberta de materiais, bem como a promessa de aceleradores disruptivos, como a computação quântica para tarefas exponencialmente caras, significa que é evidente que esses métodos se tornarão cada vez mais relevantes com o passar do tempo. É um momento apropriado para revisar as capacidades, bem como as limitações dos métodos de estrutura eletrônica subjacentes a essas simulações. Marzari, Ferretti e Wolverton abordam esta tarefa no artigo "Métodos de estrutura eletrônica para design de materiais, "acaba de ser publicado em Materiais da Natureza .
"As simulações não falham de maneiras espetaculares, mas podem mudar sutilmente de inestimáveis para apenas boas o suficiente para apenas inúteis, "disseram os autores no artigo." As razões para o fracasso são múltiplas, desde esticar as capacidades dos métodos até abandonar a complexidade dos materiais reais. Mas as simulações também são insubstituíveis:elas podem avaliar materiais em condições de pressão e temperatura tão extremas que nenhum experimento na terra é capaz de se replicar, eles podem explorar com agilidade cada vez maior o vasto espaço das fases e composições dos materiais na busca por aquela descoberta indescritível dos materiais, e podem identificar diretamente as causas microscópicas e a origem de uma propriedade macroscópica. Último, eles compartilham com todos os ramos da ciência computacional um elemento-chave de pesquisa:eles podem ser reproduzidos, abertos e compartilháveis de maneiras que nenhuma infraestrutura física jamais será. "
Os autores primeiro olham para a estrutura da teoria do funcional da densidade (DFT) e dão uma visão geral das abordagens cada vez mais complexas que podem melhorar a precisão ou estender o escopo das simulações. Eles então discutem os recursos que a ciência dos materiais computacionais desenvolveu para explorar esta caixa de ferramentas e fornecer previsões para as propriedades dos materiais sob condições realistas de complexidade cada vez maior. Finalmente, eles destacam como abordagens baseadas em física ou dados podem fornecer racionais, alto rendimento, ou avenidas de inteligência artificial para a descoberta de materiais, e explicar como esses esforços estão mudando todo o ecossistema de pesquisa.
Olhando para a frente, os autores afirmam que desenvolver métodos que possam avaliar a estabilidade termodinâmica, condições de síntese, capacidade de fabricação, e a tolerância das propriedades previstas a defeitos intrínsecos e extrínsecos em novos materiais será um desafio significativo. Os pesquisadores podem precisar aumentar as estimativas de DFT por métodos de estrutura eletrônica mais avançados ou algoritmos de aprendizado de máquina para melhorar a precisão, e usar métodos computacionais para abordar condições realistas, como entropias vibracionais, a concentração de defeitos e potenciais eletroquímicos aplicados.
Finalmente, dado o amplo papel que tais métodos provavelmente desempenharão nas próximas décadas, os autores observam que o suporte e planejamento para as infraestruturas computacionais necessárias - software científico amplamente utilizado, a verificação de códigos e validação de teorias, a disseminação e curadoria de dados computacionais, ferramentas e fluxos de trabalho, bem como os modelos de carreira associados que eles acarretam e exigem - estão apenas começando a surgir.