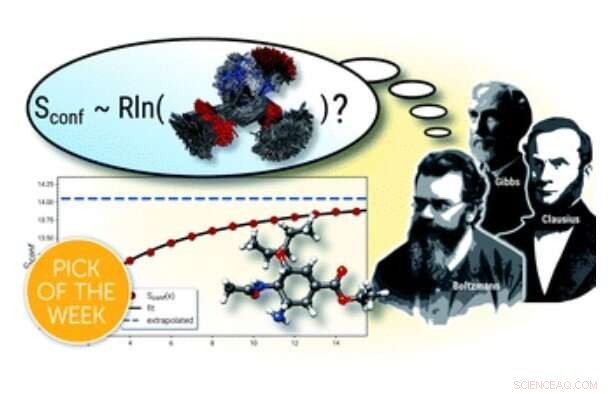
 p Resumo gráfico. Crédito:Cálculo de entropias moleculares absolutas e capacidades de calor de forma simples, Ciência Química (2021). DOI:10.1039 / D1SC00621E
p Resumo gráfico. Crédito:Cálculo de entropias moleculares absolutas e capacidades de calor de forma simples, Ciência Química (2021). DOI:10.1039 / D1SC00621E
p Químicos da Universidade de Bonn desenvolveram uma ferramenta computacional para a análise de entropias conformacionais de moléculas flexíveis. Seu método permite a investigação termodinâmica de sistemas químicos complicados pela combinação de química quântica moderna e modelos clássicos. Em uma tentativa bem-sucedida de simplificações, contribuições importantes para a entropia podem ser calculadas com intervenção mínima do usuário, mesmo em computadores desktop padrão. Os resultados são publicados na revista
Ciência Química e foram destacados como o artigo "Escolha da semana". p O termo "entropia" foi introduzido em 1865 pelo físico alemão Rudolf Clausius, que mais tarde trabalhou e foi reitor da Universidade de Bonn. 2022 será o 200º aniversário de seu aniversário e eventos científicos e celebrações estão planejados na Universidade de Bonn. A entropia é uma das propriedades termodinâmicas mais fundamentais da matéria e está comumente associada a um estado de desordem ou incerteza. Com o tempo, o conceito também se consolidou na mecânica estatística, como pioneiro pelos famosos físicos Josiah Gibbs e Ludwig Boltzmann, e na teoria da informação. Hoje, entropia é uma área ativa de pesquisa em muitos campos científicos, incluindo química computacional.
p Para as moléculas, a entropia torna-se importante como parte da descrição dependente da temperatura do interno, a chamada energia livre, do qual muitas propriedades, como equilíbrios químicos ou taxas de reação, são derivadas. Na química computacional moderna, a entropia de uma molécula é obtida a partir dos níveis de energia das vibrações atômicas dentro de uma estrutura molecular. Aqui, devido aos altos custos computacionais no nível da química quântica, várias simplificações teóricas, como a chamada aproximação de oscilador harmônico de rotor rígido, devem ser introduzidos e os cálculos são realizados principalmente para uma única estrutura. Para moléculas flexíveis, isso leva à negligência de uma importante contribuição chamada entropia conformacional, que descreve a "desordem" molecular de todas as conformações acessíveis termicamente. Esses casos flexíveis são comuns e importantes para muitas drogas farmacêuticas.
p Em uma tentativa recente de fornecer descrições dinâmicas precisas de moléculas flexíveis, O Prof. Dr. Stefan Grimme e colegas de trabalho do Centro Mulliken de Química Teórica da Universidade de Bonn desenvolveram uma nova ferramenta computacional para o cálculo de entropias conformacionais. Embora as formulações matemáticas para cálculos da entropia conformacional sejam conhecidas há algum tempo, um dos principais problemas é encontrar e avaliar o grande número de estruturas possíveis que já chegam a bilhões para moléculas de tamanho médio. Portanto, um componente central do software recém-introduzido e disponível gratuitamente é um algoritmo eficiente para esta tarefa que funciona com o mínimo de entrada do usuário, mesmo em computadores desktop padrão. Para alcançar a eficiência necessária, métodos químicos quânticos semi-empricos foram aplicados que também são desenvolvidos no grupo de Grimme, junto com cálculos de mecânica quântica padrão. No artigo foi mostrado que o procedimento é capaz de tratar até sistemas grandes e extremamente flexíveis com precisão sem precedentes para a entropia molecular. Os autores esperam que o novo protocolo computacional possa ajudar a obter dados termodinâmicos precisos de forma mais rotineira e que encontre ampla aplicação na química computacional.
p O grupo de pesquisa de Stefan Grimme trabalha com tópicos atuais da química quântica com foco na eficiência computacional e moléculas grandes. Seu colega de trabalho, Philipp Pracht, está finalizando seu doutorado. tese e é o principal autor do programa CREST empregado para os cálculos de entropia conformacional. Esta pesquisa é publicada em acesso aberto em
Ciência Química , o principal jornal revisado por pares da Royal Society of Chemistry.