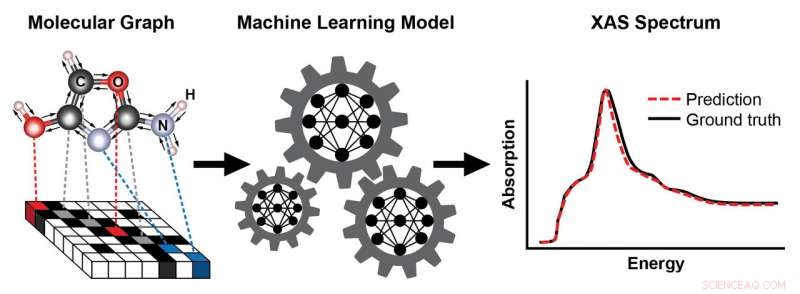
Um esquema que mostra as etapas para treinar um modelo de aprendizado de máquina para prever um espectro de absorção de raios-X (XAS) com base na estrutura conhecida de uma molécula. A estrutura da molécula é representada como um gráfico, com átomos como nós e ligações químicas como arestas. Esta representação captura a conectividade dos átomos - aqui, carbono (C), oxigênio (O), nitrogênio (N), e hidrogênio (H) - e o tipo e comprimento das ligações químicas que os conectam. O espectro XAS resultante contém informações valiosas sobre o ambiente químico local de átomos absorventes, como sua simetria e o número de átomos vizinhos. Crédito:Laboratório Nacional de Brookhaven
A espectroscopia de absorção de raios X (XAS) é uma técnica de caracterização popular para sondar a estrutura atômica local e as propriedades eletrônicas de materiais e moléculas. Como os átomos de cada elemento absorvem os raios-X em energias características, O XAS é adequado para mapear a distribuição espacial dos elementos em uma amostra. Tipicamente, cientistas realizam experimentos XAS em fontes de luz síncrotron - como a National Synchrotron Light Source II (NSLS-II) - porque eles fornecem muito brilho, raios X ajustáveis. Medindo a absorbância em uma amostra em energias de raios-X variáveis, os cientistas podem gerar um gráfico chamado espectro de absorção de raios-X.
"XAS é um recurso fundamental para usuários do NSLS-II do Brookhaven National Laboratory e do Center for Functional Nanomaterials (CFN), ambas as instalações do usuário do Office of Science do Departamento de Energia dos EUA (DOE) que estão abertas à comunidade de pesquisa científica, "disse Deyu Lu, um físico do CFN Theory and Computation Group. "Com as ferramentas de análise certas, O XAS pode fornecer insights tremendos na pesquisa em nanociência. O desenvolvimento de tais ferramentas é central para a nossa missão como facilidades do usuário. "
Classificação de ambientes químicos locais
Diferentes regiões do espectro de absorção de raios-X são sensíveis a diferentes aspectos das propriedades do material em uma amostra. Por exemplo, a estrutura de absorção de raios-X próxima à borda (XANES) concentra-se na região próxima à borda do espectro, logo acima da energia inicial suficiente para excitar um elétron das camadas internas de um átomo para um estado vazio. XANES codifica informações ricas sobre o ambiente químico local de átomos absorventes em uma amostra - incluindo sua coordenação geométrica, simetria, e estado de carga (o número de elétrons ganhos ou perdidos na ligação química). Mas analisar dados espectrais é muito desafiador por causa de sua natureza abstrata.
"Ao contrário da imagem de um microscópio de um material onde você pode ver diretamente características como cristalinidade ou defeitos, Espectros XANES codificam informações que requerem experiência de domínio para interpretar, "explicou o Lu.
A interpretação padrão de sinais em um espectro XANES se baseia em características conhecidas como "impressões digitais, "que são construídos a partir de medições em materiais de referência. No entanto, esta abordagem de impressão digital falha quando a amostra não é um cristal simples e os materiais de referência pertinentes não podem ser facilmente identificados.
Simulações em larga escala baseadas em teoria de modelos de estrutura atômica podem fornecer percepções muito úteis para a interpretação de espectros experimentais de XANES; Contudo, essas simulações costumam ser computacionalmente caras e demoradas, e seu nível de precisão depende muito das aproximações teóricas escolhidas e do sistema em estudo. Como resultado, a interpretação espectral robusta é atualmente o gargalo dos estudos XAS. Além disso, A interpretação em tempo real dos espectros XAS surgiu como um novo desafio para estudos da evolução dinâmica de materiais em condições de operação e experimentação autônoma. A necessidade de robustez, a interpretação espectral eficiente está se tornando cada vez mais difundida em fontes de luz síncrotron.
"Tempo real, interpretação precisa de espalhamento de raios-X e medidas de espectroscopia, como absorção de raios-X, fluorescência, e a difração é uma capacidade importante para os usuários que realizam pesquisas no NSLS-II e em outras instalações de luz síncrotron, "disse Mehmet Topsakal, um associado científico no Grupo de Materiais para Aplicações de Energia do Departamento de Ciência e Tecnologia Nuclear de Brookhaven, que está desenvolvendo técnicas avançadas de análise de dados e aprendizado de máquina para espectroscopia de raios-X. "Todo ano, milhares de cientistas de todo o mundo vêm ao NSLS-II para sondar as propriedades de vários materiais. Um pipeline de análise espectral de última geração permitiria aos usuários obter feedback útil sobre suas amostras enquanto os experimentos estão em andamento e fazer ajustes dinâmicos para guiar os experimentos. A questão é, como podemos fazer interpretação espectral em tempo real para descobrir correlações espectro-estrutura? "
Extração de informações com aprendizado de máquina
Aproveitando Big Data e aprendizado de máquina, Lu e Topsakal decidiram responder a essa pergunta com o cientista computacional Shinjae Yoo da Iniciativa de Ciência Computacional (CSI) do Laboratório Brookhaven e Ph.D. da Universidade de Columbia. candidato e bolsista de graduação em ciência da computação do DOE Matthew Carbone.
"O DOE Computational Science Graduate Fellowship me proporcionou uma oportunidade única de ir além da minha pesquisa de Ph.D. em física química na Universidade de Columbia para explorar o poder dos algoritmos de aprendizado de máquina, trabalhando ao lado de cientistas de Brookhaven, "disse Carbone." O aprendizado de máquina aproveita conjuntos de dados massivos para construir modelos altamente perceptivos que, uma vez treinado, pode fazer previsões instantâneas sobre novos dados. Tais modelos poderiam ser usados para contornar os caros cálculos da química quântica e auxiliar na caracterização de materiais operando. "
Os membros desta equipe e colaboradores têm trabalhado nos mapeamentos espectro-para-estrutura e estrutura-para-espectro por vários anos. Em 2017, eles desenvolveram modelos de aprendizado de máquina para prever os números médios de coordenação de nanopartículas de metal de espectros XANES. Ano passado, eles criaram um banco de dados XANES para resolver a estrutura local de um revestimento de óxido de titânio amorfo para aplicações fotocatalíticas. Eles também construíram um modelo de aprendizado de máquina capaz de prever a simetria local de átomos absorvedores de espectros simulados de XANES de óxidos de metais de transição.
"Ao realizar a interpretação espectral com base na experiência do domínio, tendemos a nos concentrar em recursos específicos criados a partir de nossa intuição, "disse Lu." O aprendizado de máquina pode extrair as informações de que precisamos de uma forma estatisticamente saliente que elimina o preconceito humano. "
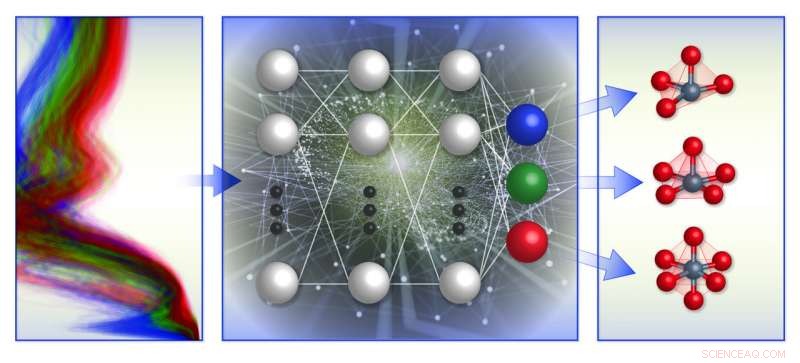
Uma ilustração esquemática da estrutura de classificação de ambiente químico local baseada em espectro da equipe. Eles treinaram modelos de aprendizado de máquina (meio) com banco de dados de espectros de absorção de raios-X computacional (esquerda) para prever a geometria local em torno de íons de metal de transição carregados positivamente (direita). Crédito:Laboratório Nacional de Brookhaven
Predizendo espectros de absorção de raios-X
Com base em seus sucessos anteriores, a equipe enfrentou um problema mais desafiador:treinar um modelo de aprendizado de máquina para prever rapidamente espectros com base em estruturas moleculares conhecidas. Tal modelo contornaria a necessidade de simulações computacionalmente caras, que não são viáveis durante experimentos operando, quando os cientistas estão estudando materiais em condições operacionais. Apesar dos esforços crescentes de aprendizado de máquina para prever as propriedades químicas dos materiais, previsões diretas das funções espectrais de materiais reais ainda não haviam sido alcançadas.
"Uma dificuldade técnica é construir uma representação ideal de estruturas moleculares que possam codificar a simetria inerente das moléculas como recursos de entrada para o modelo de aprendizado de máquina, "disse Yoo.
Adotando uma ideia recente proposta por cientistas do Google, Topsakal e Carbone construíram um modelo de aprendizado de máquina baseado em uma representação gráfica de moléculas como entrada, onde os átomos são representados como nós e ligações químicas como arestas.
"Os computadores não podem ver as moléculas como nós, "disse Topsakal." Um gráfico é uma forma natural de codificar a estrutura e conectividade de uma molécula - capturando quais átomos estão conectados e o tipo e comprimento das ligações químicas que os conectam. Além disso, esta representação é invariante para transformações como translações e rotações. Este conceito é análogo ao do reconhecimento de imagem, onde um objeto como um gato ou cachorro em um fundo ainda pode ser classificado corretamente após a imagem ser transformada. "
Para treinar o modelo para uma demonstração de prova de princípio, a equipe usou um banco de dados bem estabelecido (chamado QM9) contendo informações estruturais e químicas computadas em 134, 000 pequenas moléculas com até nove átomos pesados por tipo de átomo (carbono, azoto, oxigênio, e flúor). Deste banco de dados, eles selecionaram dois subconjuntos de treinamento - um subconjunto com moléculas contendo pelo menos um átomo de oxigênio, e outro subconjunto com moléculas contendo pelo menos um átomo de nitrogênio - e calculou seus espectros XANES correspondentes. Então, eles usaram seus modelos treinados para prever os espectros XANES para as bordas de absorção de oxigênio e nitrogênio correspondentes a excitações de elétrons na camada mais interna dos respectivos átomos.
O modelo de aprendizado de máquina reproduziu quase todos os picos de absorção significativos e previu as posições de pico (energias nas quais os picos aparecem) e alturas (intensidades de absorção) com uma precisão muito alta. O modelo também captou automaticamente o conhecimento de domínio de que a espectroscopia de absorção de raios-X é sensível a grupos funcionais, ou grupos de átomos com propriedades químicas e reatividade semelhantes. Dependendo de qual grupo funcional o átomo absorvedor pertence, características diferentes aparecem nos espectros.
"Somos os primeiros a demonstrar que um modelo de aprendizado de máquina pode ser usado para prever com precisão as funções espectrais completas de sistemas físicos reais diretamente de suas estruturas, "disse Topsakal." Embora tenhamos nos concentrado na espectroscopia de absorção de raios-X em nosso estudo, este método pode ser generalizado para prever informações espectrais para outras técnicas populares, incluindo espectroscopia infravermelha e de raios gama. "
"Depois de treinarmos o modelo de aprendizado de máquina, não precisamos executar simulações físicas demoradas, que leva minutos, horas, ou mesmo dias, "disse Yoo." Nós habilitamos não apenas a previsão de espectros em tempo real, mas também a geração simultânea de centenas e milhares de inferências de espectros usando várias unidades de processamento gráfico, ou GPUs. Essa tecnologia é a chave para permitir controles automatizados de linha de luz e acelerar as descobertas científicas. Combinado com métodos para amostrar estruturas de materiais, esses modelos podem ser usados para selecionar rapidamente estruturas relevantes para conduzir o design e a descoberta de materiais. "
Próximo, a equipe gostaria de combinar conceitos de seu modelo que prevê simetria local de espectros XANES e este novo modelo que prevê espectros XANES de estruturas moleculares. Em última análise, seu objetivo é extrair informações mais abrangentes sobre o ambiente químico local ou mesmo a estrutura de moléculas inteiras a partir de medições experimentais.
"Ferramentas de aprendizado de máquina, como aqueles para reconhecimento de imagem e fala e descoberta de drogas, estão em rápido desenvolvimento, "disse Lu." A chave é descobrir como adaptar essas ferramentas de uma forma inovadora para resolver os problemas da ciência dos materiais. "
"Nosso objetivo ao desenvolver inteligência artificial e tecnologias de aprendizado de máquina é resolver desafios científicos únicos, adotando os avanços tecnológicos mais recentes nessas áreas e criando novas abordagens que contribuam de volta para as respectivas comunidades de pesquisa, "adicionou Yoo.