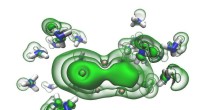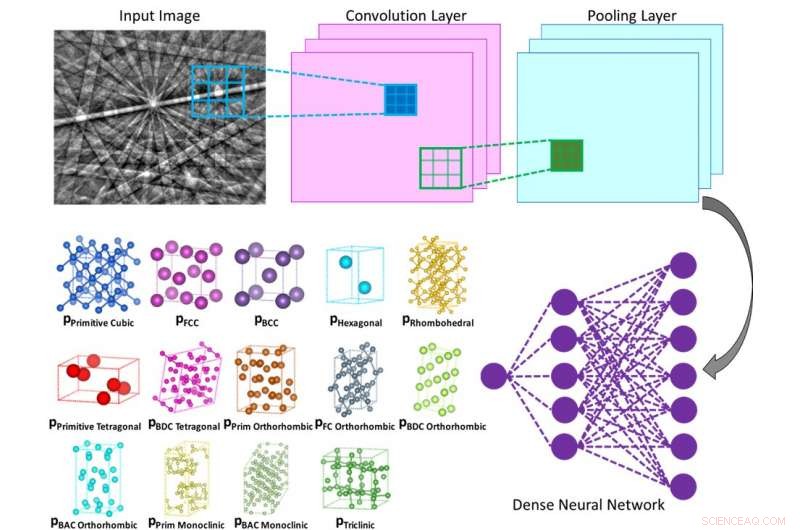
Ilustração do funcionamento interno de uma rede neural convolucional que calcula a probabilidade de que o padrão de difração de entrada pertence a uma determinada classe (por exemplo, rede de Bravais ou grupo espacial). Crédito:Laboratório Vecchio / Ciência
Os nanoengenheiros da Universidade da Califórnia em San Diego desenvolveram um método baseado em computador que pode tornar menos trabalhoso determinar as estruturas cristalinas de vários materiais e moléculas, incluindo ligas, proteínas e produtos farmacêuticos. O método usa um algoritmo de aprendizado de máquina, semelhante ao tipo usado em reconhecimento facial e carros autônomos, para analisar de forma independente os padrões de difração de elétrons, e faça isso com pelo menos 95% de precisão.
O trabalho foi publicado na edição de 31 de janeiro de Ciência .
Uma equipe liderada pelo professor de nanoengenharia da UC San Diego Kenneth Vecchio e seu Ph.D. estudante Kevin Kaufmann, quem é o primeiro autor do artigo, desenvolveu a nova abordagem. Seu método envolve o uso de um microscópio eletrônico de varredura (SEM) para coletar padrões de difração de elétrons retroespalhados (EBSD). Em comparação com outras técnicas de difração de elétrons, como aqueles em microscopia eletrônica de transmissão (TEM), EBSD baseado em SEM pode ser realizado em grandes amostras e analisadas em múltiplas escalas de comprimento. Isso fornece informações locais submícron mapeadas em escalas centimétricas. Por exemplo, um sistema EBSD moderno permite a determinação de estruturas de grãos em escala fina, orientações de cristal, tensão ou deformação residual relativa, e outras informações em uma única varredura da amostra.
Contudo, a desvantagem dos sistemas comerciais EBSD é a incapacidade do software de determinar a estrutura atômica das redes cristalinas presentes no material que está sendo analisado. Isso significa que um usuário do software comercial deve selecionar até cinco estruturas cristalinas presumivelmente na amostra e, em seguida, o software tenta encontrar correspondências prováveis para o padrão de difração. A natureza complexa do padrão de difração freqüentemente faz com que o software encontre correspondências de estrutura falsa na lista selecionada pelo usuário. Como resultado, a precisão da determinação do software existente do tipo de rede depende da experiência do operador e do conhecimento prévio de sua amostra.
O método que a equipe de Vecchio desenvolveu faz tudo isso de forma autônoma, como a rede neural profunda analisa independentemente cada padrão de difração para determinar a rede cristalina, de todos os tipos de estrutura de rede possíveis, com um alto grau de precisão (maior que 95%).
Uma ampla gama de áreas de pesquisa, incluindo farmacologia, biologia estrutural, e a geologia deve se beneficiar do uso de algoritmos automatizados semelhantes para reduzir a quantidade de tempo necessária para a identificação estrutural do cristal, pesquisadores disseram.