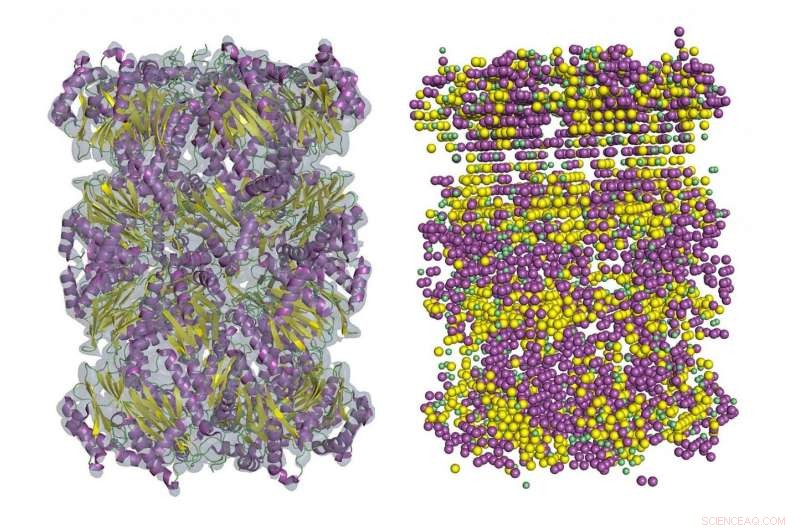
Um exemplo da detecção de estrutura secundária no mapa de densidade crio-EM usando Emap2Sec. A esquerda é um mapa EM do proteassoma arqueado 20S (EMDB ID:EMD-1733). À direita são detectadas estruturas secundárias pelo Emap2Sec. Os pontos em magenta são as posições das hélices alfa detectadas; pontos amarelos são fitas beta detectadas, e os pontos verdes são para bobinas detectadas (outras estruturas). Crédito:imagem da Universidade Purdue / Daisuke Kihara
A microscopia crioeletrônica é agora o método mais popular para determinar estruturas de proteínas, que ajuda os pesquisadores a desenvolver medicamentos para diferentes tipos de doenças. Nas últimas décadas, substituiu a cristalografia de raios-X porque pode gerar imagens de proteínas que não podem ser facilmente transformadas em grandes cristais. A nova técnica foi tão revolucionária que ganhou seus desenvolvedores o Prêmio Nobel de Química de 2017.
O produto final do crio-EM é um mapa da densidade dos átomos em moléculas biológicas, mas para atingir o nível de detalhe de que os pesquisadores precisam, eles precisam realizar uma análise mais aprofundada. Um novo estudo na revista Métodos da Natureza descreve uma técnica para trazer mapas de baixa resolução até o par.
A abordagem que os pesquisadores usam para fazer isso depende do nível de detalhe com que eles começam. Mapas de 2 a 3 ångström (Å, uma unidade de comprimento usada para expressar o tamanho de átomos e moléculas) são geralmente considerados de alta resolução. Contudo, mapas desta qualidade são difíceis de alcançar, e muitos ainda são comumente produzidos na faixa de 4 a 10 Å. De todas as proteínas depositadas no Banco de Dados de Microscopia Eletrônica de 2016-18, mais de 50% foram resolvidos na resolução intermediária.
"Se a resolução for melhor do que três, então, as ferramentas convencionais podem rastrear a posição dos aminoácidos e construir um mapa das posições dos átomos. Mas frequentemente o cryo-EM não pode lhe dar um mapa de 3 Å, "disse Daisuke Kihara, professor de ciências biológicas e ciência da computação na Purdue University. "Em mapas de 5 Å ou inferior, você geralmente não consegue ver a conectividade em cadeia. "
As proteínas são, na verdade, cadeias de aminoácidos, e a ligação entre grupos amino e grupos carboxila às vezes cria certos padrões de dobramento. Esses padrões, conhecido como alfa hélices e fitas beta, formam a estrutura secundária da proteína.
Em mapas de 5 a 8 Å, alguns fragmentos da estrutura secundária das proteínas são geralmente visíveis, mas rastrear toda a cadeia seria muito difícil. O novo método de Kihara, conhecido como Emap2sec, descobre estruturas secundárias em mapas de 6 a 10 Å.
O Emap2sec tem uma rede neural convolucional profunda no centro de seu algoritmo. Essas redes são sistemas de aprendizado profundo usados principalmente para classificar imagens, agrupe-os por similaridade e execute o reconhecimento de objetos. Ele funciona para a identificação da estrutura da proteína em mapas 3-D porque o método "convolve" os recursos de densidade do mapa local em imagens de uma região maior à medida que a informação passa pelas camadas da rede neural. A previsão local é feita no contexto de uma grande região do mapa.
Estruturas secundárias identificadas em mapas 3-D ajudam os pesquisadores a atribuir estruturas conhecidas de proteínas que já foram resolvidas no mapa. Isso significa que às vezes eles têm um ponto de partida, ou pelo menos uma pista da aparência de parte da estrutura. O Emap2sec pode ajudar os pesquisadores a encaixar sua peça no quebra-cabeça de maneira mais rápida e fácil. As informações de estrutura identificadas também podem ser úteis para localizar erros na modelagem de estrutura.