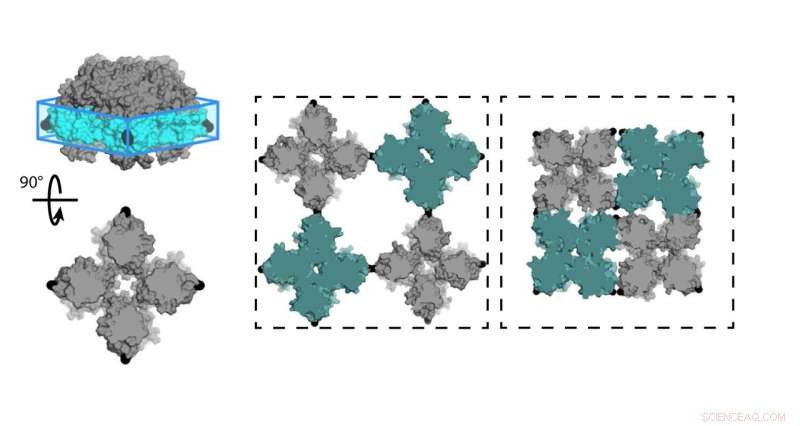
Químicos da Universidade da Califórnia, San Diego (UCSD) projetou uma folha de proteínas (C98RhuA) que alterna entre diferentes estados de porosidade e densidade. As células da estrutura cristalina são articuladas nos cantos do tetrâmero C98RhuA, permitindo que ele gire e abra ou feche o poro. Crédito:Robert Alberstein et al.
O que faz o kevlar parar uma bala, no nível atômico?
As propriedades dos materiais emergem de sua estrutura molecular ou atômica, no entanto, muitos detalhes entre o micro e o macro permanecem um mistério para a ciência. Os cientistas estão pesquisando ativamente o design racional de arquiteturas supramoleculares direcionadas, com o objetivo de projetar sua dinâmica estrutural e sua resposta aos sinais ambientais.
Uma equipe de químicos da Universidade da Califórnia, San Diego (UCSD) projetou agora um cristal de proteína bidimensional que alterna entre estados de porosidade e densidade variáveis. Este é o primeiro em design biomolecular que combinou estudos experimentais com computação feita em supercomputadores. A pesquisa, publicado em abril de 2018 em Química da Natureza , poderia ajudar a criar novos materiais para energia renovável, Medicina, purificação da água, e mais.
"Fizemos um extenso conjunto de simulações e experimentos de dinâmica molecular, que explicou a base da dinâmica estrutural incomum dessas proteínas artificiais, com base no qual fomos capazes de tomar decisões racionais e alterar a dinâmica estrutural da montagem, "disse o co-autor do estudo Akif Tezcan, professor de química e bioquímica da UCSD.
A equipe de Tezcan trabalhou com a proteína L-ramnulose-1-fosfato aldolase (RhuA), que foi modificado com aminoácidos cisteína em seus quatro cantos na posição 98 (C98RhuA). Ele e seu grupo já haviam publicado um trabalho sobre a automontagem deste artificial, arquitetura de proteína bidimensional, que ele disse ter mostrado um comportamento interessante chamado auxeticidade.
"Esses conjuntos cristalinos podem realmente abrir e fechar em coerência, "Tezcan disse." Como eles fazem, eles encolhem ou expandem igualmente nas direções X e Y, que é o oposto do que os materiais normais fazem. Queríamos investigar a que esses movimentos são devidos e o que os governa. "Um exemplo de auxeticidade pode ser visto na esfera de Hoberman, uma bola de brinquedo que se expande através de suas dobradiças em forma de tesoura quando você separa as pontas.
"Nosso objetivo era ser capaz de fazer a mesma coisa, usando proteínas como blocos de construção, para criar novos tipos de materiais com propriedades avançadas, "Disse Tezcan." O exemplo que estamos estudando aqui foi essencialmente o fruto desses esforços, onde usamos esta proteína particular que tem uma forma quadrada, que nos ligávamos um ao outro por meio de ligações químicas que eram reversíveis e agiam como dobradiças. Isso permitiu que esses materiais formassem cristais muito bem ordenados que também eram dinâmicos devido à flexibilidade dessas ligações químicas, que acabou nos dando esses novos, propriedades emergentes."
O controle da abertura e fechamento dos poros nas redes 2-D da proteína C98RhuA poderia capturar ou liberar alvos moleculares específicos úteis para a entrega de drogas ou criação de baterias melhores com mais pesquisa, Disse Tezcan. Ou eles podem passar seletivamente ou bloquear a passagem de moléculas biológicas e água filtrada.

O supercomputador Maverick é um recurso de visualização e análise de dados dedicado no Texas Advanced Computing Center, arquitetado com 132 unidades de processamento gráfico NVIDIA Tesla K40 'Atlas' (GPU) para visualização remota e computação GPU para a comunidade nacional. Crédito:TACC
"Nossa ideia era ser capaz de construir materiais complexos, como a evolução fez, usando proteínas como blocos de construção, "Disse Tezcan.
A forma como a equipe de Tezcan fez isso foi primeiro expressar as proteínas nas células da bactéria E. coli e purificá-las, após o que eles induziram a formação das ligações químicas que realmente criam os cristais de C98RhuA, que variam em função de seu estado de oxidação, através da adição de produtos químicos redox-ativos.
"Uma vez que os cristais são formados, a grande caracterização se torna a abertura ou proximidade dos próprios cristais, "explicou Tezcan, que foi determinado através da análise estatística de centenas de imagens capturadas em microscopia eletrônica.
Os experimentos trabalharam lado a lado com a computação, principalmente simulações de todos os átomos usando o software NAMD desenvolvido na Universidade de Illinois em Urbana Champaign pelo grupo do falecido biofísico Klaus Schulten.
A equipe de Tezcan usou um sistema reduzido de apenas quatro proteínas ligadas entre si, que podem ser ladrilhados infinitamente para chegar ao fundo de como o cristal abre e fecha. “O sistema reduzido nos permitiu fazer esses cálculos viáveis para nós, porque ainda existem centenas de milhares de átomos, mesmo neste sistema reduzido, "Tezcan disse. Sua equipe aproveitou recursos específicos para C98RhuA, como usar uma única coordenada de reação correspondente à sua abertura. "Fomos realmente capazes de validar este modelo como sendo representativo do que observamos no experimento, "Disse Tezcan.
As simulações moleculares de todos os átomos das redes cristalinas C98RhuA foram usadas para mapear a paisagem de energia livre. Esta paisagem de energia parece uma paisagem natural, com vales, montanhas, e passagens de montanha, explicou o co-autor do estudo Francesco Paesani, professor de química e bioquímica da UCSD.
"Os vales se tornam as configurações mais estáveis de seus conjuntos de proteínas, "Paesani disse, que o sistema molecular prefere a ter que gastar energia para subir uma montanha. E as passagens nas montanhas mostram o caminho de uma estrutura estável para outra.
"Tipicamente, cálculos de energia livre são muito caros e desafiadores porque essencialmente o que você está tentando fazer é amostrar todas as configurações possíveis de um sistema molecular que contém milhares de átomos. E você quer saber quantas posições esses átomos podem adquirir durante uma simulação. Leva muito tempo e muitos recursos de computador, "Disse Paesani.
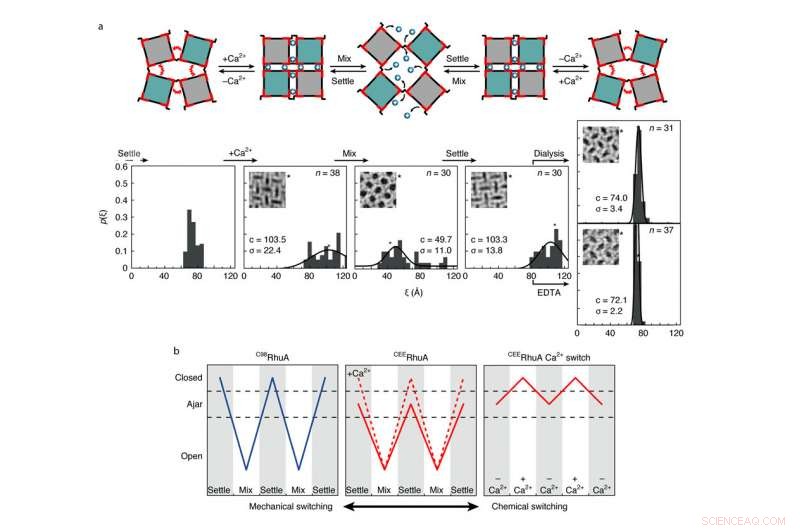
Comportamento de comutação química e mecânica de cristais CEERhuA. uma, Acima:esquemático representando todos os modos de comutação possíveis das redes CEERhuA. Abaixo:distribuição (ões) experimental (is) correspondente (s) aos estados diretamente acima. A adição de 20 mM de Ca2 + à população de equilíbrio 'entreaberta' de cristais CEERhuA induziu uma mudança em direção a conformações mais fechadas, a partir do qual a comutação mecânica semelhante ao C98RhuA foi possível. A conformação entreaberta foi totalmente recuperável após a remoção do Ca2 + por meio de diálise ou EDTA, fornecendo, assim, três modos de comutação distintos. Ajustes gaussianos para cada distribuição são rotulados com seu centro (c) e s.d. (σ). n é o número de cristais analisados. A conformação da rede de cada inserção é marcada com um asterisco. b, Resumo dos modos de comutação para cristais RhuA. Em contraste com C98RhuA, CEERhuA tem dois modos mecânicos ditados pela presença de Ca2 +, bem como um modo puramente químico por meio da adição ou remoção de Ca2 +. Crédito:Robert Alberstein et al.
Para enfrentar esses e outros desafios computacionais, Paesani recebeu alocações de supercomputador por meio do XSEDE, o Ambiente Extremo de Descoberta de Ciência e Engenharia, financiado pela National Science Foundation.
"Felizmente, XSEDE nos forneceu uma alocação no Maverick, os clusters de computação GPU no Texas Advanced Computing Center (TACC), "Paesani disse. Maverick é um recurso dedicado de visualização e análise de dados arquitetado com 132 NVIDIA Tesla K40" Atlas "unidades de processamento gráfico (GPU) para visualização remota e computação GPU para a comunidade nacional.
"Isso foi muito útil para nós, porque o software NAMD que usamos funciona muito bem em GPUs. Isso nos permite acelerar os cálculos por ordens de magnitudes, "Disse Paesani." Hoje em dia, podemos pagar cálculos com os quais há dez anos não podíamos nem sonhar por causa desses desenvolvimentos, tanto no software NAMD quanto no hardware. Todos esses clusters de computação que o XSEDE fornece são, na verdade, muito úteis para todas as simulações de dinâmica molecular. "
Por meio do XSEDE, Paesani usou vários sistemas de supercomputação, incluindo Gordon, Cometa, e cavaletes no San Diego Supercomputer Center; Kraken no Instituto Nacional de Ciências Computacionais; e Ranger, Debandada, e Stampede2 em TACC.
"Como todas as simulações foram rodadas em GPUs, Maverick foi a escolha perfeita para este tipo de aplicativo, "Disse Paesani.
A computação e a experiência trabalharam juntas para produzir resultados. "Acho que este é um belo exemplo da sinergia entre teoria e experimento, "Disse Paesani." A experiência colocou a primeira questão. Teoria e simulação de computador abordaram essa questão, fornecer alguma compreensão do mecanismo. E então usamos simulação de computador para fazer previsões e pedir aos experimentos para testar a validade dessas hipóteses. Tudo funcionou muito bem porque as simulações explicaram os experimentos no início. As previsões feitas foram confirmadas pelos experimentos no final. É um exemplo da sinergia perfeita entre experimentos e modelagem teórica. "
Tezcan acrescentou que "os químicos tradicionalmente gostam de construir moléculas complexas a partir de blocos de construção mais simples, e pode-se imaginar fazer essa combinação de design, experimento e computação para moléculas menores para prever seu comportamento. Mas o fato de que podemos fazer isso em moléculas compostas de centenas de milhares de átomos não tem precedentes. "
A equipe de ciência também usou simulações de dinâmica molecular para investigar rigorosamente o papel da água no direcionamento do movimento da rede de C98RhuA. "Este estudo nos mostrou a importância do papel ativo da água no controle da dinâmica estrutural de macromoléculas complexas, que em bioquímica pode ser esquecido, "Tezcan disse." Mas este estudo mostrou, muito claro, que a dinâmica dessas proteínas é impulsionada ativamente pela dinâmica da água, o que eu acho que traz a importância da água para o primeiro plano. "
Rob Alberstein, estudante de pós-graduação no grupo Tezcan e primeiro autor do artigo da Nature Chemistry, acrescentou "No cerne desta pesquisa está a compreensão de como as propriedades dos materiais surgem da estrutura molecular ou atômica subjacente. É muito difícil de descrever. Neste caso, realmente procuramos estabelecer essa conexão tão claramente quanto poderíamos entender nós mesmos e realmente mostrar não apenas a partir do experimento, onde podemos observar o comportamento em macroescala desses materiais, mas depois, com a computação, relacione esse comportamento com o que realmente está acontecendo na escala das moléculas. À medida que continuamos a nos desenvolver como sociedade, precisamos desenvolver novos materiais para novos tipos de problemas globais (purificação de água, etc), portanto, compreender essa relação entre a estrutura atômica e a propriedade material em si e a capacidade de predizê-las se tornará cada vez mais importante. "