Crédito:Universidade da Califórnia, São Francisco
A descoberta de medicamentos pode trazer à mente imagens de jalecos e pipetas brancos, mas quando Henry Lin, PhD, recentemente começou a encontrar um opioide melhor com menos efeitos colaterais, seu primeiro passo foi ligar os computadores.
Usando um programa chamado DOCK, ele carregou uma estrutura de cristal do receptor opióide encontrado no cérebro e acessou uma biblioteca virtual de 3 milhões de compostos que podem se ligar a uma "bolsa" química no receptor. A maioria dos medicamentos - de antibióticos a antidepressivos - funcionam ligando-se a locais específicos nas proteínas, mas para ser eficaz, eles devem se encaixar perfeitamente.
O programa girou em torno de cada composto, considerou a flexibilidade de seus vários apêndices, e depois de testar uma média de 1,3 milhão de configurações por composto - classificou-as por seu potencial de ligação. O processo, rodando em computadores conectados a processadores poderosos, levou cerca de duas semanas.
Um estudante de pós-graduação na época, Lin trabalhou com seu conselheiro Brian Shoichet, PhD, professor de química farmacêutica na Escola de Farmácia da UC San Francisco, e Aashish Manglik, PhD, da Universidade de Stanford para vasculhar os 2 primeiros, 500 compostos para fatores adicionais e 23 selecionados para testes experimentais em células vivas - jalecos de laboratório e pipetas.
Cada vez mais, pesquisadores estão recorrendo a experimentos virtuais para as etapas iniciais do desenvolvimento de medicamentos. Com computadores cada vez mais rápidos, a fase inicial e em grande parte de tentativa e erro do desenvolvimento de medicamentos pode ser reduzida a uma questão de dias, e com bibliotecas on-line cada vez maiores de compostos, triagens de drogas podem abranger, literalmente, toda a química conhecida no mundo.
Pontos fortes e limitações
Os pesquisadores estão cautelosos sobre o potencial da descoberta computacional de medicamentos - apenas uma pequena fração dos compostos promissores realmente funcionam quando testados na vida real - mas eles dizem que um de seus pontos fortes está em revelar compostos inteiramente novos como candidatos a medicamentos.
Shoichet é especialista em um método computacional popular conhecido como docking molecular. "Onde o docking se encaixa é na pesquisa de descoberta inicial, em encontrar novas partidas, " ele disse.
A busca de sua equipe pelo novo opióide ilustra os pontos fortes e as limitações da descoberta de medicamentos computacionais.
Na verdade, os candidatos a opióides iniciais identificados por meio de docking molecular tiveram um desempenho modesto em testes experimentais. "Ainda, a atividade que eles tinham era altamente reproduzível e as moléculas eram altamente novas, pressagiando uma nova biologia, "disse Shoichet.
A equipe encaixou outra rodada de compostos com estruturas semelhantes e testou os artilheiros. Com colaboradores da University of North Carolina, Chapel Hill e Friedrich Alexander University na Alemanha, eles identificaram o composto mais potente e otimizaram sua farmacologia com a elaboração sintética guiada por computador.
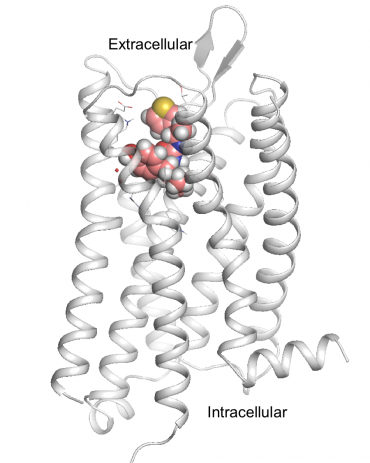
PZM21, o novo, candidato a medicamento opioide mais seguro, é mostrado encaixado no receptor de morfina do cérebro, o receptor opióide mu. Crédito:Anat Levit
Esse composto vencedor, chamado PZM21, é quimicamente diferente de qualquer outro usado atualmente e pode não ter sido encontrado por meio de métodos mais tradicionais. É um composto totalmente projetado computacionalmente que é mais potente do que a morfina. Em ratos, bloqueava a dor com eficiência, sem os efeitos colaterais usuais de supressão respiratória e constipação, e até parecia menos viciante.
Acoplar não é uma bala de prata, mas tornou-se um poderoso ponto de partida por muito tempo, processo interdisciplinar de desenvolvimento de medicamentos. Entre suas principais contribuições estão os inibidores de protease que ajudaram a tornar o HIV uma doença tratável. Os pesquisadores também estão usando docking para rastrear candidatos a medicamentos para o tratamento do câncer de mama, Hepatite C, hipertensão, Staphylococcus, o vírus SARS e a gripe.
Tecnologia pioneira na UCSF
A docagem molecular foi iniciada há três décadas por um jovem físico químico da UCSF chamado Tack Kuntz, PhD, hoje é professor emérito da Faculdade de Farmácia. Quando Kuntz chegou ao campus no início dos anos 1970, a abordagem tradicional para a descoberta de medicamentos ainda prevalecia.
Como Kuntz descreveu, o processo dependia do acaso e de muito pouca teoria:"Você sai e encontra novos compostos naturais e os traz de volta para testar em um laboratório. Basta colocar os produtos químicos junto com um organismo e ver o que acontece."
Os químicos farmacêuticos mal se preocuparam com os detalhes moleculares de como as drogas interagiam com o corpo. Muitas drogas, incluindo os primeiros antibióticos, tinha sido descoberto acidentalmente, mas Kuntz, tendo visto a nova compreensão molecular varrendo o campo da biologia, sentiu que era hora de uma atualização semelhante em farmacologia.
"A visão da biologia baseada em alvos - que você pode entender a biologia por meio de proteínas independentes e produtos genéticos - já havia assumido, mas a farmacologia estava uma década atrasada, "disse Shoichet, que foi aluno de pós-graduação no laboratório de Kuntz na década de 1980.
Kuntz e seus colegas começaram a trabalhar em direção a uma abordagem mais racional para o design de medicamentos, em que tentaram identificar compostos que pudessem se encaixar em receptores específicos nas proteínas, como encontrar a peça que faltava em um quebra-cabeça. Em 1982, eles publicaram um artigo descrevendo o primeiro programa de acoplamento molecular que poderia "explorar alinhamentos geometricamente viáveis de ligantes e receptores de estrutura conhecida."
Kuntz enviou 10, 000 exemplares desse primeiro programa de encaixe para pesquisadores de todo o país. Breve, outros pesquisadores estavam desenvolvendo programas computacionais semelhantes e o entusiasmo rapidamente se espalhou fora da academia. Na década de 1990, todas as grandes empresas farmacêuticas abriram uma unidade computacional de descoberta de medicamentos.
Pegando uma ideia
Apesar do entusiasmo inicial, Contudo, a descoberta computacional de drogas não levou a resultados rápidos. A ideia de Kuntz havia chegado antes de seu tempo. Levaria décadas de avanços incrementais em biologia molecular, tecnologia de imagem e computação, antes que a descoberta computacional de drogas pudesse começar a cumprir sua promessa.

Tack Kuntz, PhD, e seus colegas em 1982 publicaram um artigo descrevendo o primeiro programa de docking molecular que poderia “explorar alinhamentos geometricamente viáveis de ligantes e receptores de estrutura conhecida”. Crédito:Universidade da Califórnia, São Francisco
Uma limitação importante na década de 1990 foi a falta de estruturas conhecidas de proteínas. Sem estes, havia poucos alvos para os quais encontrar drogas. Nas décadas seguintes, milhares de estruturas de proteínas de possíveis alvos de drogas foram reveladas por cristalografia de raios-X e ressonância magnética nuclear.
A descoberta do novo candidato a opioide, por exemplo, só foi possível por causa das estruturas cristalinas recentemente determinadas de receptores acoplados à proteína G, uma família de proteínas que inclui o receptor opioide.
Bibliotecas virtuais de compostos também cresceram exponencialmente. Em 1991, um banco de dados pode conter 55, 000 compostos; agora eles contêm dezenas de milhões. "O escopo da química que estamos amostrando tem subido na mesma proporção da Lei de Moore, "Disse Shoichet." Há uma fome insaciável por mais e mais moléculas. "
Os programas de acoplamento de hoje são capazes de modelar de forma realista as interações em nível atômico entre uma droga e seu alvo, mas alguns detalhes complicados - como como as forças atômicas mudam quando uma molécula de droga desloca água no local de ligação - continuam sendo desafios em campo.
Promessas e Provas
A ancoragem molecular não é a única forma de design de drogas baseado em computador. No Instituto UCSF de Ciências da Saúde Computacional (ICHS), dezenas de pesquisadores estão explorando uma miríade de métodos computacionais para o avanço da pesquisa médica.
Michael Keizer, PhD, membro do ICHS e professor assistente do Instituto de Doenças Neurodegenerativas, está estudando drogas que atingem muitos alvos moleculares de uma vez, como se tocasse um acorde em vez de uma única nota. Esta ação de múltiplos alvos foi há muito entendida como a causa de efeitos colaterais não intencionais, mas também pode ser direcionado para o tratamento de doenças complexas.
Somente no início dos anos 2000 os pesquisadores reconheceram que muitos medicamentos existentes atuam em mais de um alvo - os antipsicóticos, por exemplo, que atingiu os receptores de serotonina e dopamina. Eles agora estão projetando drogas intencionalmente para isso.
“Para algumas doenças que ainda não têm tratamento, talvez seja porque não há uma única proteína que você precise ativar ou desativar; e se a droga precisar atingir vários alvos em vez disso? ", disse Keizer, que era um estudante graduado de Shoichet.
Em seu laboratório, Keizer usa métodos computacionais para identificar padrões químicos entre drogas que se ligam ao mesmo conjunto de alvos e encontrar novos compostos que possuem farmacologia compatível. Esta abordagem computacional pode reconhecer semelhanças entre compostos que as análises mais convencionais perderiam. Keizer agora está procurando por tecnologia de inteligência artificial, conhecido como aprendizado profundo, para um reconhecimento de padrões ainda melhor.
Mesmo com os métodos computacionais decolando, sua prova ainda está no mundo real - em células, modelos animais, e, finalmente, na clínica. “Por um tempo, era comum publicar artigos com previsões sobre as atividades de uma pequena molécula, mas nenhum teste real dessas previsões, porque os experimentos para fazer isso eram caros, difícil ou esotérico, "disse Keizer.
Como a necessidade de colaboração se tornou clara, a parceria entre previsão computacional e experimentos de laboratório úmido se fortaleceu visivelmente na última década, disse Keizer. "Afinal, como você pode melhorar suas previsões se não tem certeza de quais estão erradas? "