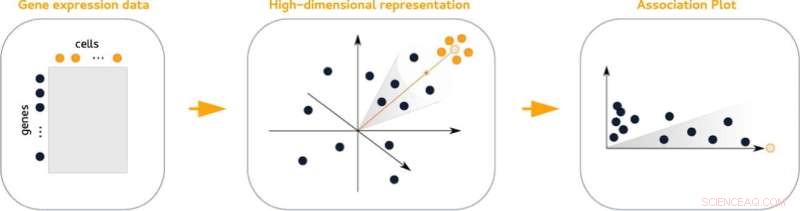
Resumo gráfico. Crédito:Jornal de Biologia Molecular (2022). DOI:10.1016/j.jmb.2022.167525
Quais genes são específicos para um determinado tipo de célula, ou seja, "marcam" sua identidade? Com o tamanho crescente dos conjuntos de dados hoje em dia, responder a essa pergunta geralmente é um desafio. Muitas vezes, os genes marcadores são simplesmente genes que foram encontrados em populações celulares específicas. No entanto, muitos outros genes podem ser característicos de um tipo de célula específico, mas permanecem desconhecidos.
Gráficos de associação (APL), um novo método estatístico para visualizar a atividade gênica dentro de um aglomerado de células, facilitam a localização de seus genes marcadores. Os gráficos comparam a atividade dos genes de um determinado cluster com todos os outros clusters do conjunto de dados. Além disso, eles facilitam a visualização de quais genes são compartilhados com outros clusters.
"Os gráficos de associação não apenas nos permitem identificar novos genes marcadores. Também funciona ao contrário - somos capazes de combinar grupos de identidade desconhecida em um conjunto de dados para tipos de células, com base em uma lista fornecida de genes marcadores", diz Elzbieta Gralinska do Instituto Max Planck de Genética Molecular em Berlim.
O biotecnólogo trabalha na equipe de Martin Vingron, que desenvolveu a técnica. Os pesquisadores demonstraram a funcionalidade da técnica em dois conjuntos de dados disponíveis publicamente e publicaram os resultados no
Journal of Molecular Biology . Além disso, o APL foi lançado como um módulo gratuito para o ambiente estatístico R. O pacote APL permite que os pesquisadores inspecionem visualmente seus dados de célula única e selecionem genes individuais com o cursor para aprender mais detalhes.
Analisando e agrupando células únicas Por que é necessário identificar genes marcadores em primeiro lugar? As modernas tecnologias de sequenciamento são capazes de decifrar moléculas de RNA individuais em células individuais. A partir de uma amostra de sangue, por exemplo, cada célula pode ser separada e uma amostra dos RNAs da célula pode ser decodificada. Esses dados de célula única representam os genes ativos que foram transcritos em moléculas de RNA.
A vantagem:em vez de se perguntar a qual tipo de célula um RNA específico pertence, ele pode ser rastreado até sua célula de origem. A desvantagem:sequenciar milhares de RNAs em cada célula de dezenas de milhares de células produz quantidades extraordinárias de dados.
Uma saída é classificar as células com base em seu conteúdo de RNA. "Os dados de uma única célula são compostos de uma mistura selvagem de muitos tipos de células diferentes. Estamos interessados em células do mesmo tipo de célula, que devem se comportar de forma semelhante", explica Martin Vingron. Portanto, faz sentido agrupar células semelhantes computacionalmente, diz ele. "Para nós, os genes marcadores definem um tipo de célula."
Explorando clusters de células interativamente Usando dados publicamente disponíveis de glóbulos brancos, a equipe demonstrou como o novo algoritmo funciona. Os muitos tipos diferentes de glóbulos brancos, como células T, células B ou monócitos, são agrupados em grupos separados. Os pesquisadores confirmaram genes marcadores conhecidos e foram capazes de mostrar que parentes próximos entre as células do sangue também compartilham grande semelhança em sua atividade genética.
"Cada um dos genes marcadores que encontramos com APL poderia ter sido descoberto por pelo menos um outro método existente para identificação de genes marcadores", diz Gralinska. Mas a vantagem do APL sobre os algoritmos existentes é sua representação gráfica dos resultados, diz ela. "As ferramentas existentes fornecem longas listas de genes e valores de pontuação. Muitas vezes, os usuários percorrem a lista e param em um corte arbitrário."
Por outro lado, o novo método oferece uma maneira de visualizar esses genes, clicar em cada um e observar mais de perto sua atividade, diz ela. "Não estamos apenas fornecendo listas de genes marcadores, estamos permitindo que os usuários analisem como esses genes se comportam", diz o pesquisador. “Com gráficos de associação, eles podem mergulhar em seus dados para aprender mais sobre cada tipo de célula”. Além disso, diz ela, é muito fácil decompor o papel biológico dos genes mais interessantes em uma etapa subsequente por meio da análise de enriquecimento dos termos da Gene Ontology, que é compatível com o software APL - algo que ela considera "um recurso muito útil".
O modelo matemático subjacente Os dados de alta dimensão que contêm informações sobre a atividade entre os genes não podem ser representados visualmente sem perda de informações. O mesmo vale para dados agrupados, o que complica a análise. "Nosso truque é levar em conta muito mais do que apenas duas ou três dimensões, mas, em última análise, criamos um diagrama bidimensional", diz Gralinska.
Os gráficos de associação são derivados de uma técnica matemática que incorpora simultaneamente genes e células em um espaço comum de alta dimensão. Medir as distâncias entre os genes e um determinado cluster de células neste espaço resulta em pares de valores que refletem a associação de um gene a um determinado cluster e fornecem informações sobre sua associação com outros clusters.
"Uma deficiência do APL é que contamos com dados pré-agrupados, o que significa que temos que confiar em outras técnicas de agrupamento", diz Martin Vingron. "No entanto, esperamos que nosso novo método encontre muitos novos usuários. Achamos que um processo visual e interativo simplesmente faz uma análise melhor."