Shantenu Jha, presidente do Brookhaven Lab's Center for Data-Driven Discovery, e sua equipe da Rutgers University e University College London projetou uma estrutura de software para calcular com precisão e rapidez com que força os candidatos a medicamentos se ligam a suas proteínas-alvo. A estrutura visa resolver o problema do mundo real do design de medicamentos - atualmente um processo longo e caro - e pode ter um impacto na medicina personalizada. Crédito:Laboratório Nacional de Brookhaven
Soluções para muitos problemas científicos e de engenharia do mundo real - desde o aprimoramento de modelos climáticos e o projeto de novos materiais de energia até a compreensão de como o universo se formou - exigem aplicativos que podem ser dimensionados para um tamanho muito grande e de alto desempenho. Cada ano, por meio de seu Desafio Internacional de Computação Escalável (ESCALA), o Instituto de Engenheiros Elétricos e Eletrônicos (IEEE) reconhece um projeto que avança no desenvolvimento de aplicativos e infraestrutura de suporte para permitir a expansão, computação de alto desempenho necessária para resolver esses problemas.
O vencedor deste ano, "Enabling trade-off between Accuracy and Computational Cost:Adaptive Algorithms to Reduce Time to Clinical Insight, "é o resultado de uma colaboração entre químicos e cientistas da computação e da computação no Laboratório Nacional de Brookhaven do Departamento de Energia dos EUA (DOE), Universidade Rutgers, e University College London. Os membros da equipe foram homenageados no 18º IEEE / Association for Computing Machinery (ACM) Simpósio Internacional sobre Cluster, Computação em nuvem e grade realizada em Washington, DC, de 1 a 4 de maio.
"Desenvolvemos uma metodologia de computação numérica para avaliar com precisão e rapidez a eficácia de diferentes candidatos a medicamentos, "disse o membro da equipe Shantenu Jha, presidente do Center for Data-Driven Discovery, parte da Iniciativa de Ciência da Computação do Brookhaven Lab. "Embora ainda não tenhamos aplicado esta metodologia para desenvolver um novo medicamento, demonstramos que poderia funcionar em grandes escalas envolvidas no processo de descoberta de drogas. "
A descoberta de drogas é como projetar uma chave para caber em uma fechadura. Para que um medicamento seja eficaz no tratamento de uma doença específica, ele deve se ligar fortemente a uma molécula - geralmente uma proteína - que está associada a essa doença. Só então a droga pode ativar ou inibir a função da molécula alvo. Os pesquisadores podem selecionar 10, 000 ou mais compostos moleculares antes de encontrar qualquer um que tenha a atividade biológica desejada. Mas esses compostos "principais" muitas vezes não têm a potência, seletividade, ou estabilidade necessária para se tornar uma droga. Ao modificar a estrutura química dessas ligações, os pesquisadores podem criar compostos com as propriedades semelhantes às de drogas. Os candidatos a medicamentos projetados, então, passam ao longo do pipeline de desenvolvimento para o estágio de teste pré-clínico. Desses candidatos, apenas uma pequena fração entra na fase de ensaio clínico, e apenas um acaba se tornando um medicamento aprovado para uso pelo paciente. Levar um novo medicamento ao mercado pode levar uma década ou mais e custar bilhões de dólares.
Superando gargalos de design de medicamentos por meio da ciência da computação
Avanços recentes em tecnologia e conhecimento resultaram em uma nova era de descoberta de medicamentos - uma era que poderia reduzir significativamente o tempo e as despesas do processo de desenvolvimento de medicamentos. Melhorias em nossa compreensão das estruturas cristalinas 3-D de moléculas biológicas e aumentos no poder de computação estão tornando possível o uso de métodos computacionais para prever interações droga-alvo.
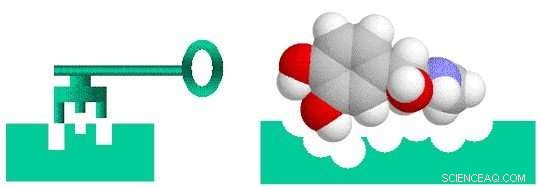
A descoberta de um medicamento é um problema de bloqueio e chave no qual o medicamento (chave) deve se ajustar especificamente ao alvo biológico (bloqueio). Crédito:Laboratório Nacional de Brookhaven
Em particular, uma técnica de simulação de computador chamada dinâmica molecular tem se mostrado promissora em prever com precisão a força com que as moléculas de drogas se ligam a seus alvos (afinidade de ligação). A dinâmica molecular simula como os átomos e as moléculas se movem conforme interagem em seu ambiente. No caso da descoberta de drogas, as simulações revelam como as moléculas de drogas interagem com sua proteína alvo e mudam a conformação da proteína, ou forma, que determina sua função.
Contudo, esses recursos de previsão ainda não estão operando em escala grande ou velocidade suficiente para que as empresas farmacêuticas os adotem em seu processo de desenvolvimento.
"Traduzir esses avanços em precisão preditiva para impactar a tomada de decisão industrial requer que na ordem de 10, 000 afinidades de ligação são calculadas o mais rápido possível, sem a perda de precisão, "disse Jha." A produção de uma visão oportuna exige uma eficiência computacional baseada no desenvolvimento de novos algoritmos e sistemas de software escaláveis, e a alocação inteligente de recursos de supercomputação. "
Jha e seus colaboradores na Rutgers University, onde também é professor do Departamento de Engenharia Elétrica e de Computação, e a University College London projetou uma estrutura de software para oferecer suporte ao cálculo preciso e rápido de afinidades de ligação e, ao mesmo tempo, otimizar o uso de recursos computacionais. Esta estrutura, chamada Calculadora de Afinidade de Ligação de Alta Taxa de Transferência (HTBAC), baseia-se no projeto RADICAL-Cybertools que Jha lidera como investigador principal do Laboratório de Pesquisa em Infraestrutura Cibernética Distribuída Avançada e Aplicações (RADICAL) da Rutgers. O objetivo do RADICAL-Cybertools é fornecer um conjunto de blocos de construção de software para suportar os fluxos de trabalho de aplicativos científicos de grande escala em plataformas de computação de alto desempenho, que agregam poder de computação para resolver grandes problemas computacionais que, de outra forma, seriam insolúveis devido ao tempo necessário.
Na ciência da computação, fluxos de trabalho referem-se a uma série de etapas de processamento necessárias para concluir uma tarefa ou resolver um problema. Especialmente para fluxos de trabalho científicos, é importante que os fluxos de trabalho sejam flexíveis para que possam se adaptar dinamicamente durante o tempo de execução para fornecer os resultados mais precisos enquanto fazem uso eficiente do tempo de computação disponível. Esses fluxos de trabalho adaptativos são ideais para a descoberta de medicamentos porque apenas os medicamentos com altas afinidades de ligação devem ser avaliados posteriormente.
"O trade-off desejado entre a precisão necessária e o custo computacional (tempo) muda ao longo da descoberta da droga conforme o processo passa da triagem para a seleção do lead e, em seguida, para a otimização do lead, "disse Jha." Um número significativo de compostos deve ser rastreado de forma barata para eliminar ligantes pobres antes que métodos mais precisos sejam necessários para discriminar os melhores ligantes. Fornecer o tempo de solução mais rápido requer monitorar o progresso das simulações e basear as decisões sobre a execução contínua em significância científica. "
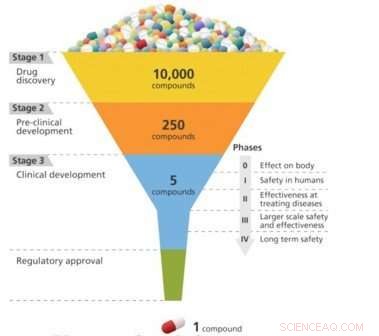
Um esquema do processo de desenvolvimento de medicamentos, que progressivamente atinge os candidatos mais eficazes de um grande pool inicial. Crédito:Laboratório Nacional de Brookhaven
Em outras palavras, não faria sentido continuar as simulações de uma interação droga-proteína em particular se a droga se ligasse fracamente à proteína em comparação com os outros candidatos. Mas faria sentido alocar recursos computacionais adicionais se um medicamento mostrar uma alta afinidade de ligação.
O suporte a fluxos de trabalho adaptativos em grandes escalas, característicos de programas de descoberta de medicamentos, requer recursos computacionais avançados. O HTBAC fornece esse suporte por meio de uma camada de software de middleware flexível que permite a execução adaptativa de algoritmos. Atualmente, O HTBAC oferece suporte a dois algoritmos:amostragem aprimorada de dinâmica molecular com aproximação de solvente contínuo (ESMACS) e integração termodinâmica com amostragem aprimorada (TIES). ESMACS, um método computacionalmente mais barato, mas menos rigoroso do que TIES, calcula a força de ligação de um medicamento à sua proteína alvo com base em simulações de dinâmica molecular. Por contraste, TIES compara as afinidades de ligação relativas de dois medicamentos diferentes à mesma proteína.
"ESMACS fornece uma abordagem quantitativa rápida e sensível o suficiente para determinar as afinidades de ligação para que possamos eliminar ligantes fracos, enquanto TIES fornece um método mais preciso para investigar bons ligantes à medida que são refinados e melhorados, "disse Jumana Dakka, um Ph.D. de segundo ano aluno da Rutgers e membro do grupo RADICAL.
Para determinar qual algoritmo executar, HTBAC analisa os cálculos de afinidade de ligação em tempo de execução. Essa análise informa as decisões sobre o número de simulações simultâneas a serem realizadas e se as etapas de estimulação devem ser adicionadas ou removidas para cada candidato a medicamento investigado.
Colocando a estrutura à prova
Jha's team demonstrated how HTBAC could provide insight from drug candidate data on a short timescale by reproducing results from a collaborative study between University College London and the London-based pharmaceutical company GlaxoSmithKline to discover drug compounds that bind to the BRD4 protein. Known to play a key role in driving cancer and inflammatory diseases, the BRD4 protein is a major target of bromodomain-containing (BRD) inhibitors, a class of pharmaceutical drugs currently being evaluated in clinical trials. The researchers involved in this collaborative study are focusing on identifying promising new drugs to treat breast cancer while developing an understanding of why certain drugs fail in the presence of breast cancer gene mutations.
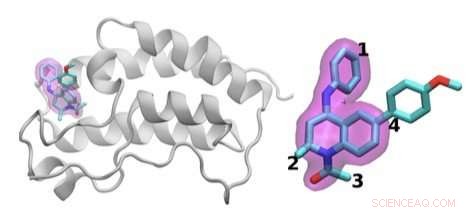
The scientists investigated the chemical structures of 16 drugs based on the same tetrahydroquinoline (THQ) scaffold. On the left is a cartoon of the BRD4 protein bound to one of these drugs; on the right is a molecular representation of a drug with the THQ scaffold highlighted in magenta. Regions that are chemically modified between the drugs investigated in this study are labeled 1 to 4. Typically, only a small change is made to the chemical structure of one drug to the next. This conservative approach makes it easier for researchers to understand why one drug is effective, whereas another is not. Crédito:Laboratório Nacional de Brookhaven
Jha and his team concurrently screened a group of 16 closely related drug candidates from the study by running thousands of computational sequences on more than 32, 000 computing cores. They ran the computations on the Blue Waters supercomputer at the National Center for Computing Applications, University of Illinois at Urbana-Champaign.
In a real drug design scenario, many more compounds with a wider range of chemical properties would need to be investigated. The team members previously demonstrated that the workload management layer and runtime system underlying HTBAC could scale to handle 10, 000 concurrent tasks.
"HTBAC could support the concurrent screening of different compounds at unprecedented scales—both in the number of compounds and computational resources used, " said Jha. "We showed that HTBAC has the ability to solve a large number of drug candidates in essentially the same amount of time it would take to solve a smaller set, assuming the number of processors increases proportionally with the number of candidates."
This ability is made possible through HTBAC's adaptive functionality, which allows it to execute the optimal algorithm depending on the properties of the drugs being investigated, improving the accuracy of the results and minimizing compute time.
"The lead optimization stage usually considers on the order of 10, 000 small molecules, " said Jha. "While experiment automation reduces the amount of time needed to calculate the binding affinities, HTBAC has the potential to cut this time (and cost) by an order of magnitude or more."
With HTBAC, TIES requires approximately 25, 000 central processing unit (CPU) core hours for a single prediction. At least a 250 million core hours would be needed for a large-scale study to support a pharmaceutical drug screening campaign, with a typical turnaround time of about two weeks. HTBAC could facilitate running studies requiring sustained usage of millions of core hours per day.
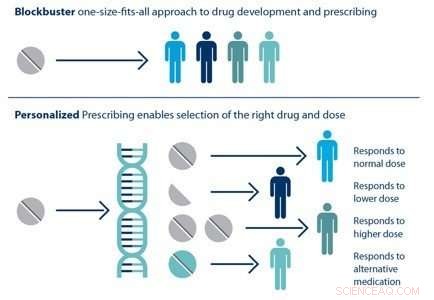
Individual patients respond differently to drugs. The ability to predict which treatment is best for a particular patient based on his or her genetic sequence is the goal of personalized medicine. Crédito:Laboratório Nacional de Brookhaven
When the University of College London–GlaxoSmithKline study concludes, Jha and his team hope to be given the experimental data on the tens of thousands of drug candidates, without knowing which candidate ended up being the best one. Com esta informação, they could perform a blind test to determine whether HTBAC provides an improvement in compute time (for a given accuracy) over the existing automated methods for drug discovery. Se necessário, they could then refine their methodology.
Applying scalable computing to precision medicine
HTBAC not only has the potential to improve the speed and accuracy of drug discovery in the pharmaceutical industry but also to improve individual patient outcomes in clinical settings. Using target proteins based on a patient's genetic sequence, HTBAC could predict a patient's response to different drug treatments. This personalized assessment could replace the traditional one-size-fits-all approach to medicine. Por exemplo, such predictions could help determine which cancer patients would actually benefit from chemotherapy, avoiding unnecessary toxicity.
According to Jha, the computation time would have to be significantly reduced in order for physicians to clinically use HTBAC to treat their patients:"Our grand vision is to apply scalable computing techniques to personalized medicine. If we can use these techniques to optimize drugs and drug cocktails for each individual's unique genetic makeup on the order of a few days, we will be empowered to treat diseases much more effectively."
"Extreme-scale computing for precision medicine is an emerging area that CSI and Brookhaven at large have begun to tackle, " said CSI Director Kerstin Kleese van Dam. "This work is a great example of how technologies we originally developed to tackle DOE challenges can be applied to other domains of high national impact. We look forward to forming more strategic partnerships with other universities, pharmaceutical companies, and medical institutions in this important area that will transform the future of health care."