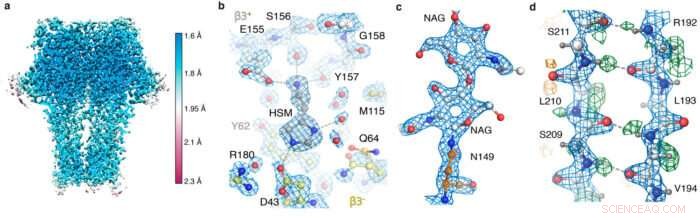
GABA UMA instantâneos do mapa do receptor. (a) resolução local; (b) a bolsa do agonista mostrando a coordenação da histamina e as moléculas de água; (c) glicano ligado a N; (d) rede de ligações de hidrogênio revelada pelo mapa de diferença (picos verdes).
Olhar para o arranjo tridimensional preciso dos átomos dentro de uma proteína nos ajuda a entender como ela pode desempenhar suas funções. Embora a crio-microscopia eletrônica (crio-EM) tenha se desenvolvido rapidamente como uma importante técnica de biologia estrutural nos últimos anos, A cristalografia de raios X foi a única técnica capaz de visualizar átomos individuais. Grupos de Radu Aricescu e Sjors Scheres no Laboratório MRC de Biologia Molecular, em colaboração com cientistas da Thermo Fisher Scientific e de outros lugares, agora foram capazes de resolver átomos de proteínas individuais pela primeira vez em uma imagem crio-EM tridimensional.
Essa colaboração começou no início de 2019 quando Radu e Abhay Kotecha, um pesquisador da Thermo Fisher Scientific, queria testar um novo hardware crio-EM em uma pequena amostra de proteína de membrana. Receptores GABAA, um foco da pesquisa de Radu por mais de uma década, foram escolhidos porque a maior resolução alcançável usando a melhor tecnologia disponível parecia ter atingido um limite em torno de 2,5 Ångströms (Å), mas uma resolução mais alta era claramente necessária para um melhor design de drogas.
O que é resolução atômica?
A resolução é geralmente relatada em Ångströms, uma unidade de comprimento que é um décimo bilionésimo de um metro ou 0,1 nanômetro, e refere-se à menor distância entre a qual dois objetos podem ser vistos como separados.
O comprimento de uma ligação carbono-carbono típica é 1,5 Å; outras ligações nas proteínas são um pouco mais curtas. Assim, conforme a resolução cai para 1,2 Å, torna-se possível ver átomos individuais dentro de uma proteína, alcançar a verdadeira resolução atômica.
Enquanto testava novos desenvolvimentos de hardware que incluíam uma fonte de elétrons de canhão de emissão de campo frio, um novo filtro de energia, e uma nova câmera, a equipe também teve que desenvolver novas estratégias de processamento. Algoritmos para a correção de aberrações ópticas que foram desenvolvidos anteriormente por Jasenko Zivanov no grupo de Sjors, bem como um algoritmo proposto por Chris Russo e Richard Henderson, desempenhou um papel crucial em extrair o máximo de informações das imagens.
Depois de receber as imagens coletadas no novo hardware do microscópio por Abhay Kotecha da Thermo Fisher Scientific em Eindhoven, Holanda, Takanori Nakane, um pós-doutorado no grupo de Sjors, desenvolveu um fluxo de trabalho ideal no RELION e Andrija Sente, junto com outros membros do grupo de Radu, usou este fluxo de trabalho para processar imagens do receptor GABAA, enquanto informa os resultados para otimizar rapidamente as configurações do microscópio. Um novo, O sistema de armazenamento de dados de alta capacidade desenvolvido por Jake Grimmett e Toby Darling na equipe de Computação Científica do LMB ofereceu suporte crucial para lidar com os cerca de cem terabytes de dados gerados. Esse esforço sustentado da equipe levou a uma estrutura de receptor GABAA de resolução de 1,7 Å sem precedentes.
Esta foi a melhor resolução relatada alcançada usando crio-EM para qualquer amostra de proteína diferente da proteína apoferritina. A apoferritina é comumente usada como referência para crio-EM, porque sua estabilidade molecular e simetria de 24 vezes permitem reconstruções de alta resolução a partir de relativamente poucas partículas.
Usando o novo hardware e estratégias de processamento, a equipe foi capaz de obter uma estrutura de apoferritina com resolução de 1,22 Å, batendo o recorde anterior de 1,53 Å para ser a estrutura crio-EM de partícula única de mais alta resolução já obtida. Mais impressionante, esta resolução permitiu a visualização de átomos de hidrogênio individuais, até mesmo nas moléculas de água dentro da estrutura da proteína. A visualização de redes de ligações de hidrogênio dentro de estruturas de proteínas e em bolsas de ligação de drogas permite aos pesquisadores entender melhor como elas funcionam.
Este trabalho representa a quebra de uma barreira fundamental para crio-EM como uma técnica de biologia estrutural e a nova tecnologia, coleção de dados, e as estratégias de processamento irão expandir o número de proteínas cujas estruturas podem ser resolvidas em alta resolução. Essas reconstruções de alta resolução permitirão um melhor entendimento de como as proteínas funcionam e facilitarão o projeto de drogas mais específicas que podem impactar no tratamento de uma grande variedade de doenças.