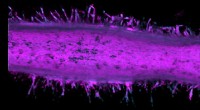
p O aprendizado de máquina prevê a estrutura e dinâmica das nanopartículas Nanoestruturas, como essas nanopartículas de ouro cobertas por tiol, agora podem ser estudadas usando o novo método de aprendizado de máquina desenvolvido na Universidade de Jyväskylä. O método pode prever a energia potencial de uma determinada estrutura de forma confiável. Crédito:Antti Pihlajamäki / The University of Jyväskylä
p O aprendizado de máquina prevê a estrutura e dinâmica das nanopartículas Nanoestruturas, como essas nanopartículas de ouro cobertas por tiol, agora podem ser estudadas usando o novo método de aprendizado de máquina desenvolvido na Universidade de Jyväskylä. O método pode prever a energia potencial de uma determinada estrutura de forma confiável. Crédito:Antti Pihlajamäki / The University of Jyväskylä
p Pesquisadores do Centro de Nanociência e da Faculdade de Tecnologia da Informação da Universidade de Jyväskylä na Finlândia demonstraram que os novos métodos de aprendizado de máquina à distância desenvolvidos na Universidade de Jyväskylä são capazes de prever estruturas e dinâmicas atômicas de nanopartículas de forma confiável. Os novos métodos são significativamente mais rápidos do que os métodos de simulação tradicionais usados para pesquisa de nanopartículas e facilitarão explorações mais eficientes das reações partícula-partícula e da funcionalidade das partículas em seu ambiente. O estudo foi publicado em uma edição especial dedicada ao aprendizado de máquina no
Journal of Physical Chemistry em 15 de maio, 2020. p Os novos métodos foram aplicados a nanopartículas de metal estabilizadas com ligante, que foram estudados por muito tempo no Centro de Nanociência da Universidade de Jyväskylä. Ano passado, os pesquisadores publicaram um método que é capaz de prever com sucesso os locais de ligação das moléculas de ligante estabilizantes na superfície das nanopartículas. Agora, uma nova ferramenta foi criada que pode prever com segurança a energia potencial com base na estrutura atômica da partícula, sem a necessidade de usar cálculos de estrutura eletrônica numericamente pesados. A ferramenta facilita as simulações de Monte Carlo da dinâmica do átomo das partículas em temperaturas elevadas.
p A energia potencial de um sistema é uma quantidade fundamental na nanociência computacional, uma vez que permite avaliações quantitativas da estabilidade do sistema, taxas de reações químicas e forças de ligações interatômicas. Nanopartículas de metal estabilizadas por ligante têm muitos tipos de ligações interatômicas de força química variável, e tradicionalmente as avaliações de energia têm sido feitas usando a chamada teoria do funcional da densidade (DFT) que freqüentemente resulta em cálculos numericamente pesados que requerem o uso de supercomputadores. Isso impediu simulações eficientes para compreender as funcionalidades das nanopartículas, por exemplo., como catalisadores, ou interações com objetos biológicos, como proteínas, vírus, ou DNA. Métodos de aprendizado de máquina, uma vez treinado para modelar os sistemas de forma confiável, pode acelerar as simulações em várias ordens de magnitude.
p
O novo método permitiu que as simulações fossem executadas em um laptop ou desktop
p Neste trabalho, os pesquisadores usaram as energias potenciais, previsto pelo método de aprendizado de máquina, para simular a dinâmica atômica de nanopartículas de ouro estabilizadas com tiol. Os resultados estão de acordo com as simulações realizadas com o uso da teoria do funcional da densidade. O novo método permitiu que as simulações fossem executadas em um laptop ou desktop em uma escala de tempo de algumas horas, enquanto as simulações DFT de referência levavam dias em um supercomputador e usavam simultaneamente centenas ou até milhares de núcleos de computador. A aceleração permitirá simulações de longo tempo das mudanças estruturais das partículas e reações partícula-partícula em temperaturas elevadas.
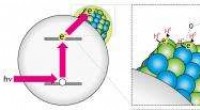
p Os pesquisadores usaram um método de aprendizado de máquina à distância desenvolvido no grupo do professor Tommi Kärkkäinen em Jyväskylä. Ele descreve cada configuração atômica momentânea de uma nanopartícula por meio do cálculo de um descritor, e compara distâncias entre descritores em um espaço numérico multidimensional. Usando correlações com um conjunto de treinamento criado pelas simulações DFT de referência, a energia potencial pode ser prevista. Esta abordagem, usado agora pela primeira vez na pesquisa de nanopartículas, é mais simples e mais transparente do que as redes neurais usadas tradicionalmente.
p "É extremamente motivador podermos reduzir a carga computacional da execução de simulações em supercomputadores para executá-las com qualidade semelhante em um laptop ou PC doméstico, "diz o estudante de doutorado Antti Pihlajamäki, que é o principal autor do estudo.
p "Foi uma grande surpresa que nossos métodos de aprendizado de máquina relativamente simples funcionem tão bem para nanoestruturas complicadas, "afirma o professor Tommi Kärkkäinen.
p “Na próxima fase, nosso objetivo é generalizar o método para funcionar bem com nanopartículas de diversos tamanhos e composições químicas. Ainda precisaremos de supercomputadores para gerar dados de alta qualidade suficientes para treinar o algoritmo de aprendizado de máquina, mas esperamos que no futuro possamos passar a usar esses novos métodos principalmente para estudos de funcionalidade de nanopartículas em ambientes químicos complicados, "resume o professor da Academia Hannu Häkkinen, quem coordenou o estudo.