 p Stuart Lindsay é o diretor do Center for Single Molecule Biophysics do Biodesign Institute da Arizona Arizona State University. Crédito:The Biodesign Institute da Arizona State University
p Stuart Lindsay é o diretor do Center for Single Molecule Biophysics do Biodesign Institute da Arizona Arizona State University. Crédito:The Biodesign Institute da Arizona State University
p Cerca de três bilhões de pares de bases constituem o genoma humano - a planta baixa da vida. Em 2003, o Projeto Genoma Humano anunciou a descriptografia bem-sucedida deste código, um tour de force que continua a fornecer uma série de percepções relevantes para a saúde e doenças humanas. p No entanto, os principais atores em praticamente todos os processos vitais são as proteínas codificadas por sequências de DNA conhecidas como genes. Para um amplo espectro de doenças, proteínas podem render revelações muito mais convincentes do que podem ser colhidas apenas do DNA, se os pesquisadores conseguirem desbloquear as sequências de aminoácidos das quais são compostos.
p Agora, Stuart Lindsay e seus colegas do Instituto de Biodesign da Universidade do Estado do Arizona deram um grande passo nessa direção, demonstrando a identificação precisa de aminoácidos, fixando brevemente cada um em uma junção estreita entre um par de eletrodos flanqueadores e medindo uma cadeia característica de picos de corrente passando por moléculas de aminoácidos sucessivas.
p Usando um algoritmo de aprendizado de máquina, Lindsay e sua equipe foram capazes de treinar um computador para reconhecer surtos de atividade elétrica que representam a ligação momentânea de um aminoácido dentro da junção. Os sinais de ruído foram mostrados para agir como impressões digitais confiáveis, identificação de aminoácidos, incluindo variantes sutilmente modificadas.
p As proteínas já estão fornecendo uma grande quantidade de informações pertinentes a doenças, incluindo câncer, diabetes e distúrbios neurológicos como Alzheimer, além de fornecer informações importantes sobre outro processo mediado por proteínas:o envelhecimento.
p O novo trabalho avança a perspectiva de sequenciamento de proteínas clínicas e a descoberta de novos biomarcadores - faróis de alerta precoce que sinalizam doenças. Avançar, o sequenciamento de proteínas pode transformar radicalmente o tratamento do paciente, permitindo o monitoramento preciso da resposta da doença à terapêutica, no nível molecular.
p Os resultados da pesquisa do grupo são relatados na edição online avançada da revista
Nature Nanotechnology .
p
Do genoma ao proteoma
p Uma enorme biblioteca de proteínas, conhecida como proteoma, ocupa o centro do palco em praticamente todos os processos vitais. As proteínas são vitais para o crescimento celular, diferenciação e reparo; eles catalisam reações químicas e fornecem defesa contra doenças, entre uma miríade de funções de limpeza.
p Uma das surpresas mais estranhas que surgiram do Projeto Genoma Humano é o fato de que apenas cerca de 1,5% do genoma codifica proteínas. O resto dos nucleotídeos de DNA formam sequências regulatórias, genes de RNA não codificantes, introns, e DNA não codificador, (uma vez chamado de "DNA lixo" zombeteiramente). Isso deixa os humanos com escassos 20-25, 000 genes, uma descoberta preocupante, visto que a lombriga humilde tem aproximadamente o mesmo número. Como observa a professora Lindsay, a notícia fica pior:"Um lírio tem cerca de uma ordem de magnitude mais genes do que nós, " ele diz.
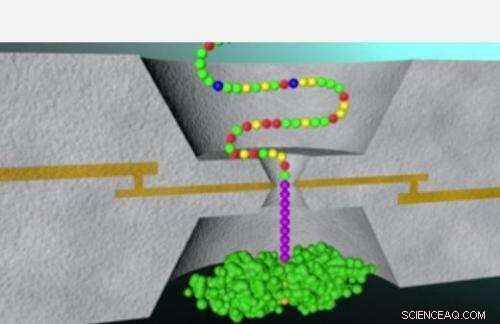
Esta animação mostra o processo básico de sequenciamento de aminoácidos em um nanopore, usando a técnica de tunelamento de reconhecimento. Crédito:The Biodesign Institute da Arizona State University p O mistério de organismos complexos como os humanos com um número de genes assustadoramente baixo tem a ver com o fato de que as proteínas geradas a partir do projeto de DNA podem ser modificadas de várias maneiras. Na verdade, os cientistas já identificaram mais de 100, 000 proteínas humanas e pesquisadores como Lindsay acreditam que isso pode ser apenas a ponta do iceberg.
p Assim como as frases podem ter seus significados alterados por meio de mudanças na ordem das palavras ou pontuação, proteínas geradas a partir de modelos de genes podem mudar de função (ou, às vezes, tornar-se inoperantes), frequentemente com consequências graves para a saúde humana. Dois processos-chave que modificam proteínas são conhecidos como splicing alternativo e modificação pós-tradução. Eles são os condutores da extraordinária variação proteica observada.
p O splicing alternativo ocorre ao codificar regiões de RNA, (conhecidos como exons) são unidos e regiões não codificantes (conhecidas como íntrons) são cortadas, antes da tradução em proteínas. Este processo nem sempre ocorre perfeitamente, com sobreposições ocasionais de exons ou introns sendo introduzidos, produzindo proteínas com splicing alternativo, cuja função pode ser alterada.
p Modificações pós-tradução são marcadores adicionados após as proteínas terem sido feitas. Existem muitas formas de modificação pós-tradução, incluindo metilação e fosforilação. Algumas proteínas alteradas realizam funções vitais, enquanto outros podem ser aberrantes e associados a doenças (ou propensão a doenças). Vários tipos de câncer estão associados a esses erros de proteínas, que já são usados como marcadores de diagnóstico. A identificação adequada de tais proteínas, entretanto, permanece um grande desafio na biomedicina.
p
Novas sequências
p A técnica descrita na pesquisa atual foi aplicada anteriormente no laboratório de Lindsay para o sequenciamento bem-sucedido de bases de DNA. Esse método, conhecido como tunelamento de reconhecimento, envolve a passagem de um peptídeo por um pequeno orifício conhecido como nanopore. Um par de eletrodos de metal, separados por um intervalo de aproximadamente dois nanômetros, fica em cada lado do nanoporo conforme unidades sucessivas de um peptídeo são enfiadas através da pequena abertura, com cada unidade completando um circuito elétrico e emitindo uma explosão de picos de corrente.
p O grupo de pesquisa demonstrou que análises detalhadas desses picos atuais podem permitir aos pesquisadores determinar qual das quatro bases de nucleotídeo - adenina, timina, citosina ou guanina - estava posicionada entre os eletrodos no nanoporo.
p "Há cerca de 2 anos, em uma de nossas reuniões de laboratório, foi sugerido que talvez a mesma tecnologia funcionasse para aminoácidos, "Lindsay diz. Assim, começaram os esforços para enfrentar o desafio substancialmente maior de usar o tunelamento de reconhecimento para identificar todos os 20 aminoácidos encontrados nas proteínas, em oposição a apenas 4 bases compreendendo DNA.
p O sequenciamento de proteínas de uma única molécula é de enorme valor, offering the potential to detect diminishingly small quantities of proteins that may have been tweaked by alternative splicing or post-translational modification. Muitas vezes, these are the very proteins of interest from the standpoint of recognizing disease states, though current technologies are inadequate to detect them.
ASU Regents' Professor Stuart Lindsay discusses the use of nanotechnology for next-generation protein and DNA sequencing and its potential impact on personalized medicine. Credit:Ken Fagan, Arizona State University p As Lindsay notes, there is no equivalent in the protein world to polymerase chain reaction (PCR) technology, which allows minute quantities of DNA in a sample to be rapidly amplified. "We probably don't even know about most of the proteins that would be important in diagnostics. It's just a black hole to us because the concentrations are too low for current analytical techniques, " ele diz, adding that the ability of recognition tunneling to pinpoint abnormalities on a single molecule basis "could be a complete game changer in proteomics."
p The new paper describes a series of experiments in which pure samples of individual amino acids, individual molecules in mixed solution and finally, short peptide chains were successfully identified through recognition tunneling. The work sets the stage for a method to sequence individual protein molecules rapidly and cheaply (see accompanying animation).
p A machine learning algorithm known as Support Vector Machine was used to train a computer to analyze the burst signals produced when amino acids formed bonds in the tunnel junction and emitted a lively noise signal as the poised electrodes passed tunneling current through each molecule. (The machine learning algorithm is the same one used by the IBM computer 'Watson' to defeat a human opponent in Jeopardy.)
p Lindsay says that around 50 distinct signal burst characteristics were used in the amino acid identifications, but that most of the discriminatory power is achieved with 10 or fewer signal traits.
p Remarkably, recognition tunneling not only pinpointed amino acids with high reliability from single complex burst signals, but managed to distinguish a post-translationally modified protein (sarcosine) from its unmodified precursor (glycine) and also to discriminate between mirror-image molecules knows as enantiomers and so-called isobaric molecules, which differ in peptide sequence but exhibit identical masses.
p
Pathway to the $1000 dollar proteome?
p Lindsay indicates that the new studies, which rely on innovative strategies for handling single molecules coupled with startling advances in computing power, open up horizons that were inconceivable only a short time ago. It is becoming clear that the tools that made the $1000 genome feasible are equally applicable to an eventual $1000 dollar proteome. De fato, such a landmark may not be far off. "Por que não?" Lindsay asks. "People think it's crazy but the technical tools are there and what will work for DNA sequencing will work for protein sequencing."
p While the tunneling measurements have until now been made using a complex laboratory instrument known as a scanning tunneling microscope (STM), Lindsay and his colleagues are currently working on a solid state device capable of fast, cost-effective and clinically applicable recognition tunneling of amino acids and other analytes. Eventual application of such solid-state devices in massively parallel systems should make clinical proteomics a practical reality.