Pesquisadores do Laboratório Nacional Argonne do Departamento de Energia dos EUA (DOE) desenvolveram uma nova técnica para acelerar cálculos que revelam como os elétrons interagem nos materiais. Este avanço permitirá aos cientistas projetar novos materiais para uma variedade de aplicações, incluindo células solares, baterias e catalisadores.
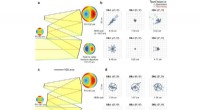
A nova técnica, chamada de "teoria do funcional de densidade de campo autoconsistente com triagem dinâmica" (SCF-DFT + DS), reduz o custo computacional de cálculo da densidade eletrônica em um material em até 90% em comparação aos métodos convencionais. Isto torna possível realizar cálculos em sistemas muito maiores, como aqueles encontrados em materiais do mundo real.
"SCF-DFT+DS é um avanço significativo no campo da ciência dos materiais", disse a cientista de Argonne, Giulia Galli, que liderou a equipe de pesquisa. "Isso nos permitirá estudar uma gama mais ampla de materiais e fenômenos e projetar novos materiais com propriedades aprimoradas para uma variedade de aplicações."
A técnica SCF-DFT+DS é baseada em uma reformulação das equações da teoria do funcional da densidade (DFT). DFT é um método amplamente utilizado para calcular a estrutura eletrônica de materiais, mas pode ser computacionalmente caro para sistemas grandes. A nova técnica utiliza uma representação simplificada da interação elétron-elétron, o que reduz o custo computacional sem sacrificar a precisão.
A equipe de pesquisa testou a nova técnica em vários sistemas, incluindo semicondutores, metais e isolantes. Eles descobriram que SCF-DFT+DS produziu resultados que estavam em excelente concordância com a DFT convencional, mas a uma fração do custo computacional.
"SCF-DFT+DS é uma ferramenta nova e poderosa que abrirá novas possibilidades para pesquisa de materiais", disse Galli. “Estamos entusiasmados em explorar seu potencial e usá-lo para projetar novos materiais para um futuro mais limpo e sustentável”.
A pesquisa foi publicada na revista Physical Review Letters.