Nova técnica pode tornar a modelagem de moléculas muito mais fácil
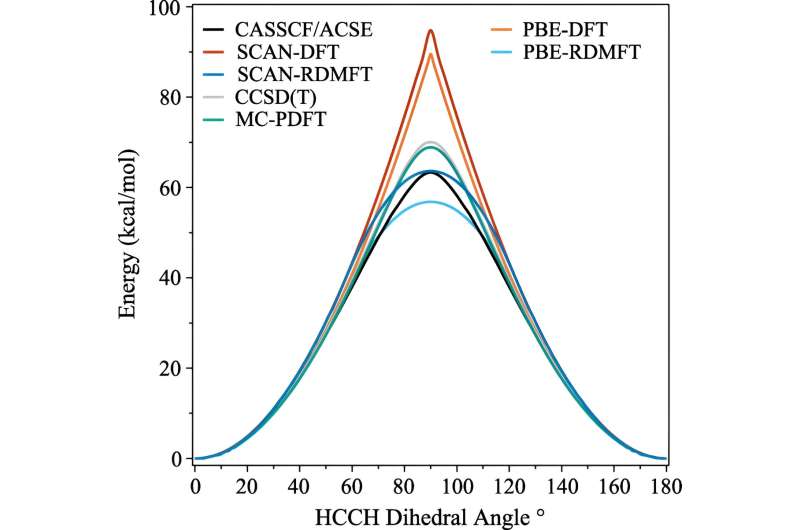
C2 H4 superfícies de energia potencial de barreira rotacional obtidas de cálculos CASSCF(12,12)/ACSE, CCSD(T), PBE-RDMFT, SCAN-RDMFT, PBE-DFT, SCAN-DFT e CASSCF(12,12)/tPBE com o cc -conjunto de base pVDZ. Crédito:Cartas de revisão física (2023). DOI:10.1103/PhysRevLett.131.243003 Assim como os humanos que os criaram, os computadores consideram a física difícil, mas a mecânica quântica é ainda mais difícil. Mas uma nova técnica criada por três cientistas da Universidade de Chicago permite que os computadores simulem certos efeitos desafiadores da mecânica quântica em materiais eletrônicos complexos com muito menos esforço.
Ao tornar estas simulações mais precisas e eficientes, os cientistas esperam que a técnica possa ajudar a descobrir novas moléculas e materiais, como novos tipos de células solares ou computadores quânticos.
“Este avanço tem um imenso potencial para aprofundar a nossa compreensão dos fenómenos moleculares, com implicações significativas para a química, ciência dos materiais e campos relacionados”, disse o cientista Daniel Gibney, Ph.D. estudante de química e primeiro autor do artigo, publicado em 14 de dezembro na Physical Review Letters .
Elétrons e energia
Uma folha ou um painel solar parece suave e simples visto de fora, mas aproxime-se do nível molecular e você verá uma dança extremamente complicada de elétrons e moléculas.
Para fazer novos avanços na sustentabilidade, na indústria, na agricultura e em muitos outros campos, os cientistas modelam o comportamento destas interações químicas e moleculares. Isto ajuda a revelar novas possibilidades de design para o futuro – para tudo, desde novas formas de sequestrar dióxido de carbono até novos tipos de bits quânticos.
Muitos avanços foram feitos nas últimas décadas, mas uma das áreas que permaneceu teimosamente difícil de simular é quando as moléculas começam a exibir comportamentos mecânicos quânticos complexos que os cientistas chamam de forte correlação.
O problema é que, quando os elétrons começam a exibir seus efeitos mais mecânicos quânticos – como ficarem “emaranhados” – os cálculos precisam instantaneamente de muito mais poder computacional. Até os supercomputadores lutam para lidar com as implicações.
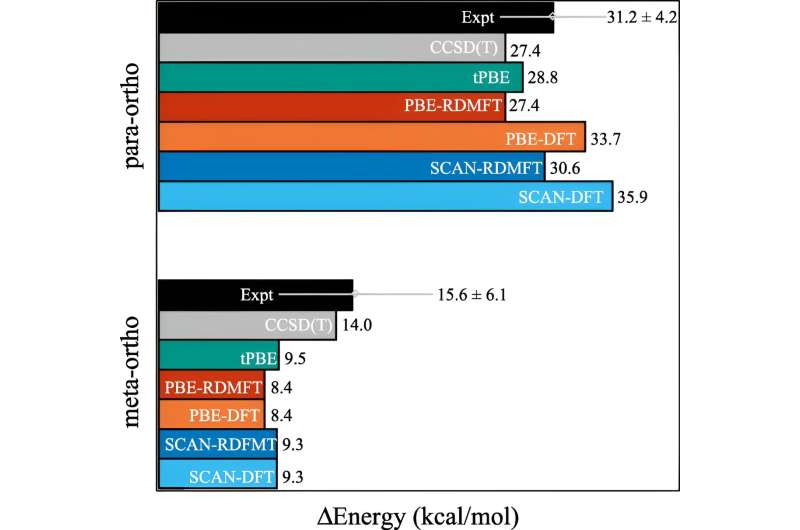
Um dos cálculos comumente usados é chamado de teoria do funcional da densidade. “Esta é basicamente a técnica mais onipresente para prever a estrutura eletrônica, mas é essencialmente uma aproximação onde todos os elétrons são tratados como uma função de um elétron”, explicou David Mazziotti, professor de química e autor sênior do estudo. Energias relativas de meta- e para-benzina em relação ao orto-benzina de RDMFT e DFT com os funcionais SCAN e PBE, MC-PDFT usando o funcional tPBE e CCSD(T). Crédito:Cartas de revisão física (2023). DOI:10.1103/PhysRevLett.131.243003 Para muitos cálculos, uma aproximação resolve o problema. Mas começa a desmoronar à medida que o comportamento dos eletrões se torna mais correlacionado, como acontece quando a mecânica quântica começa a entrar em ação. Na mecânica quântica, esses elétrons podem estar em vários lugares, ou orbitais, simultaneamente. Isso frustra não apenas os cérebros humanos, mas também a teoria do funcional da densidade.
"E este é um problema importante, porque muitas das questões que nos preocupam no século 21 - como novas moléculas e materiais para energia renovável e sustentabilidade - exigem que exploremos a natureza quântica dos materiais", disse Mazziotti.
Mazziotti, Gibney e o terceiro autor, Jan-Niklas Boyn, descobriram que poderiam adicionar uma correção universal à teoria do funcional da densidade que permite que os elétrons fiquem emaranhados entre vários orbitais ao mesmo tempo.
“Isso permite que os orbitais no cálculo não sejam apenas totalmente preenchidos ou totalmente vazios, mas em qualquer lugar intermediário”, disse Mazziotti. "Chegamos a uma imagem de um elétron que ainda é capaz de capturar o comportamento que surge dos efeitos correlacionados de elétrons de muitos corpos."
Uma adaptação 'universal'
Como bônus, disseram os cientistas, o código pode ser adicionado a algoritmos existentes sem a necessidade de reescrever esse código. “Basicamente, a correção entra em ação sempre que necessária, mas não interfere no resto do código”, disse Gibney.
É também universal – na medida em que pode ser adicionado a códigos que simulam muitos tipos de comportamento eletrónico, sejam painéis solares fotovoltaicos, sequestro de carbono ou materiais supercondutores – ou mesmo biologia.
Por exemplo, explicou Boyn, uma aplicação poderia ser a compreensão da química que ocorre usando enzimas contendo átomos de metal, conhecidas como metaloenzimas.
“Há uma infinidade de metaloenzimas responsáveis por grande parte da química das células, por exemplo, mas elas têm sido notoriamente difíceis de descrever com os modelos atuais”, disse ele. "Esta teoria poderá, num futuro próximo, permitir-nos abordar esta química de uma forma que é impossível neste momento."
Mais informações: Daniel Gibney et al, Teoria do Funcional da Generalização Universal da Densidade para Correlação Estática, Physical Review Letters (2023). DOI:10.1103/PhysRevLett.131.243003 Informações do diário: Cartas de revisão física