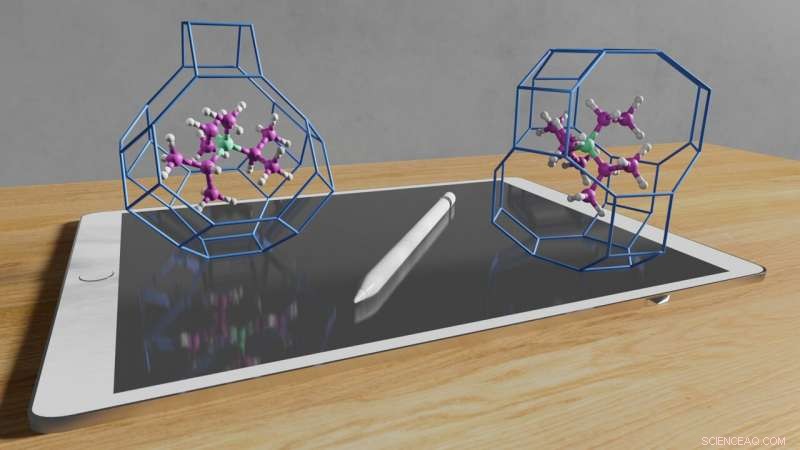
Os métodos computacionais permitem controlar a competição de fase entre estruturas zeólitas. Impressão artística de como uma molécula projetada por computador é capaz de sintetizar duas estruturas diferentes. Crédito:Schwalbe-Koda et al.
Zeólitos, grupos de minerais compreendendo aluminossilicatos hidratados, são conhecidos por serem materiais altamente promissores para uma série de aplicações. Por exemplo, eles podem ser usados como catalisadores, trocadores de cátions e peneiras moleculares.
Embora muitos estudos anteriores tenham examinado o potencial desses materiais, Até agora, o gerenciamento da competição de fase durante a síntese do zeólito provou ser desafiador e trabalhoso. O termo síntese de zeólitos se refere aos processos pelos quais os zeólitos podem ser criados ou sintetizados em laboratório.
Pesquisadores do Instituto de Tecnologia de Massachusetts (MIT), em colaboração com pesquisadores da Universidade Politécnica de Valência e da Universidade de Estocolmo, propuseram recentemente uma nova estratégia para controlar a seletividade de fase durante os processos de síntese de zeólita modelada. Esta estratégia, apresentado em um artigo publicado em Ciência , é baseado no uso combinado de simulações atomísticas, mineração de literatura, interações homem-computador, técnicas de síntese e caracterização de materiais.
"Nossa pesquisa no Learning Matter Lab do MIT se concentra em problemas de agulha no palheiro na ciência dos materiais, “Rafael Gomez-Bombarelli, um dos pesquisadores que realizou o estudo, disse a Phys.org. "Projetar uma molécula que molda seletivamente um determinado zeólito tem sido um problema combinatório difícil por décadas, com muitas tentativas e erros no laboratório. Embora as simulações atomísticas tenham ajudado historicamente, as abordagens tradicionais não tinham o papel da seletividade porque se concentravam em um único zeólito por vez. "

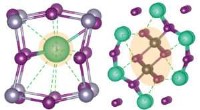
Os métodos computacionais permitem o projeto de modelos para cavidades de zeólita. Crédito:Schwalbe-Koda et al.
Gomez-Bombarelli e seus colegas usaram simulações de alto rendimento baseadas na mecânica molecular para quantificar a afinidade de diferentes modelos moleculares para o zeólito que estavam tentando criar e aqueles que eram inadequados para uma determinada aplicação. A equipe obteve informações de mais de 586, 000 simulações de moléculas de zeólita que foram alinhadas com a literatura existente em design de materiais.
"Usando essas simulações, encontramos modelos que são mais seletivos, mesmo que não sejam os ligantes mais fortes, "Daniel Schwalbe-Koda, outro pesquisador envolvido no estudo, disse a Phys.org. "Graças aos algoritmos rápidos que ajustamos no ano anterior e que comparamos com décadas de dados da literatura, nossas simulações foram ordens de magnitude mais rápidas do que as abordagens tradicionais e nos permitiram alcançar um grande número de combinações de forma muito eficiente. "
Os resultados das simulações levaram à identificação de vários projetos possíveis para zeólitas que poderiam ser realizados no futuro. Embora não haja certeza de que todos os designs que eles identificaram seriam ideais, o trabalho de Gomez-Bombarelli, Schwalbe-Koda e seus colegas podem ajudar a restringir a busca por designs de zeólitos promissores e acelerar os processos de síntese de zeólitos.
"A teoria normalmente apoiou experimentos na ciência dos zeólitos, mas raramente lidera o caminho, "Manuel Moliner, um dos pesquisadores que conduziu o estudo, disse a Phys.org. "Com esses novos insights, nossas chances de sucesso quando nos propomos a fazer novos materiais no laboratório são muito maiores e há muito potencial inexplorado em moléculas que não receberam atenção, mas podem desbloquear novos, catalisadores eficientes e econômicos. "
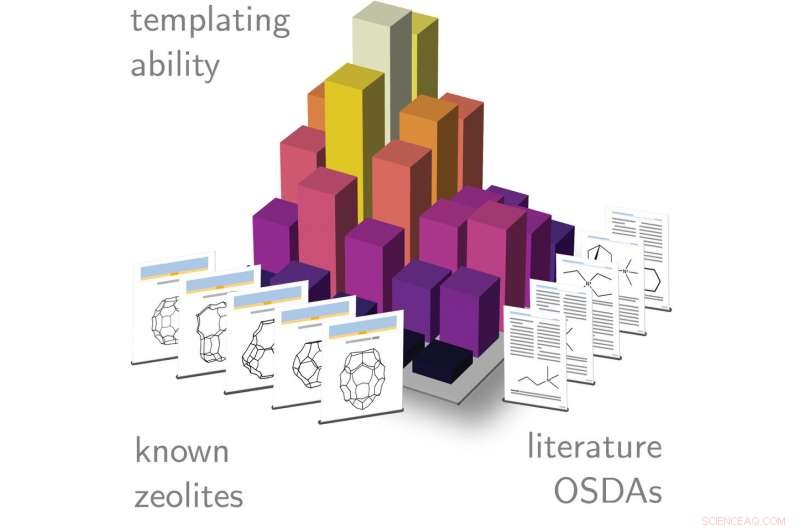
A competição de fase é quantificada para mais de 200 zeólitas conhecidas e todos os modelos da literatura. Os resultados da simulação permitem aos pesquisadores encontrar não apenas moléculas boas para uma determinada estrutura, mas quais estruturas são susceptíveis de cristalizar quando uma determinada molécula é usada na síntese. Crédito:Schwalbe-Koda et al.
Este estudo recente confirma que algoritmos e ferramentas computacionais de alto desempenho podem desempenhar um papel fundamental na identificação de novos materiais promissores. Apesar disso, os pesquisadores acreditam fortemente que a intuição de humanos especialistas ainda é necessária ao analisar simulações de computador ou as previsões de um algoritmo.
"No fim do dia, humanos são os usuários finais dos dados, portanto, devemos tentar torná-lo o mais útil possível para aplicações práticas, "Schwalbe-Koda disse." Um dos meus insights favoritos de nosso estudo é que a forma molecular é um grande preditor de seletividade. Conseguimos criar um novo material que está a meio caminho entre dois já conhecidos, usando um modelo cuja forma está a meio caminho entre as moléculas tradicionalmente usadas. "
A nova estratégia computacional para controlar a síntese de zeólitas e composição de estrutura apresentada por Gómez-Bombarelli, Schwalbe-Koda, Moliner e seus colegas poderão em breve ajudar na descoberta de novos modelos de zeólita promissores. Isso pode ter implicações importantes para vários campos de pesquisa, incluindo o campo da energia e os esforços para enfrentar as alterações climáticas. Os pesquisadores decidiram, portanto, disponibilizar seus dados publicamente por meio de um site interativo online.
"Existem muitos caminhos interessantes para pesquisas futuras, "Moliner disse." Dois que são de interesse teórico e prático vêm à mente. Um é personalizar a composição e a geometria da bolsa catalítica no zeólito e avançar para "enzimas inorgânicas". Outra é descobrir zeólitas completamente novas que por enquanto permanecem puramente hipotéticas. Ao disponibilizar nossos dados de simulação para a comunidade, esperamos que outros também sejam inspirados a buscar novas direções criativas. "
© 2021 Science X Network