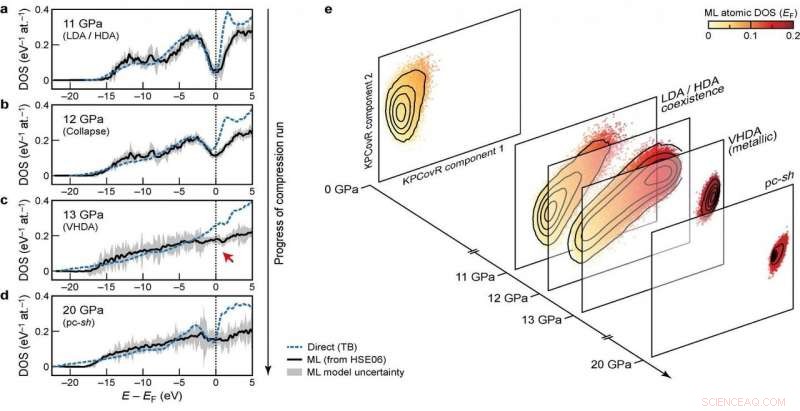
Densidades eletrônicas de estados (DOS) em vários estágios da execução de compressão. Crédito:@Michele Ceriotti
Combinar cálculos de estrutura eletrônica e técnicas de aprendizado de máquina (ML) tornou-se uma abordagem comum na modelagem atomística da matéria. Usar as duas técnicas juntas permitiu aos pesquisadores, por exemplo, para criar modelos que usam coordenadas atômicas como as únicas entradas para prever de forma econômica qualquer propriedade que possa ser computada pelos cálculos dos primeiros princípios que foram usados para treiná-los.
Embora os esforços mais antigos e agora mais avançados tenham se concentrado no uso de previsões de energias totais e forças atômicas para construir potenciais interatômicos, esforços mais recentes têm como alvo propriedades adicionais de cristais e moléculas, como energias de ionização, Proteções químicas NMR, propriedades de resposta dielétrica e densidade de carga. No artigo "Aprendendo a densidade eletrônica dos estados na matéria condensada, "Ceriotti e colegas se concentram na densidade eletrônica de estados (DOS), outra quantidade subjacente a muitas propriedades de materiais úteis, alguns dos quais podem ser observados experimentalmente.
O DOS é essencialmente o número de diferentes estados que os elétrons podem ocupar em um determinado nível de energia, e pode ser usado, por exemplo, calcular a contribuição eletrônica para a capacidade de calor em metais e a densidade de portadores de carga livre em semicondutores. É um proxy indireto para propriedades como o gap de energia, a banda de energia e o espectro de absorção óptica.
"Prever o DOS é um exercício interessante por si só porque é essencialmente a descrição mais simples possível da estrutura eletrônica além da imagem do estado fundamental, "Ceriotti disse." Também é útil porque há muitas propriedades que você pode calcular a partir do DOS, tornando-o um ótimo exemplo de como a próxima geração de modelos de ML pode ser usada de maneira semelhante aos cálculos de estrutura eletrônica, usá-los de forma indireta para calcular quantidades intermediárias que podem então ser facilmente processadas para avaliar propriedades que são mais difíceis de aprender diretamente. "
Ao desenvolver o modelo, o grupo procurou garantir a capacidade de transferência em diferentes fases, bem como a escalabilidade para sistemas de grande porte. Sua abordagem final, que analisa como as diferentes configurações atômicas afetam a distribuição dos níveis de energia, atende a esses objetivos - foi capaz de aprender e prever o DOS computado por DFT para um conjunto de dados diversificado de estruturas de silício, cobrindo uma ampla gama de condições termodinâmicas e diferentes fases. Ele também é dimensionado linearmente, em vez do cubo do número de átomos, como nos cálculos de estrutura eletrônica, tornando-o aplicável a grandes estruturas. Finalmente, o modelo permitiu uma análise do DOS local, dando aos pesquisadores a chance de examinar a interação entre os motivos estruturais e a estrutura eletrônica.
A combinação de transferibilidade, e escalabilidade de previsões para grandes tamanhos de sistema, tornar o modelo aplicável para tratar de questões em aberto de longa data na ciência dos materiais. A nova estrutura já foi usada para elucidar as propriedades eletrônicas de uma simulação de silício amorfo de 100.000 átomos, passando por uma série de transições de fase quando comprimido a 20 Gpa, em um artigo publicado em Natureza hoje em colaboração com uma equipe composta por pesquisadores de Oxford, Cambridge, o Laboratório de Pesquisa Naval dos EUA e a Universidade de Ohio. O DOS previsto também é usado para explicar como as transformações estruturais induzidas por pressão se acoplam à estrutura eletrônica do material.
Combinar o novo modelo com um dos modelos de energia potencial bem estabelecidos também torna possível calcular as contribuições eletrônicas para propriedades macroscópicas, como a capacidade térmica de metais e realizar simulações que levam em consideração efeitos de temperatura eletrônicos finitos - como demonstrado em outro artigo a ser publicado discutindo as propriedades do níquel em alta temperatura. De fato, o novo modelo é uma etapa crítica em direção ao objetivo da MARVEL de desenvolver modelos integrados de aprendizado de máquina que aumentam - e talvez eventualmente substituam - os caros cálculos de estrutura eletrônica.
"Existem outras propriedades além da densidade de elétrons dos estados, como excitações ópticas, e resposta NMR, que pudemos prever com precisão com o aprendizado de máquina ". Ceriotti disse." Se pudermos usá-los todos em combinação com potenciais interatômicos baratos e precisos, isso nos permitirá descrever todas as propriedades dos materiais com a mesma precisão alcançada com a eletrônica cálculo da estrutura, mas por uma pequena fração do custo. "