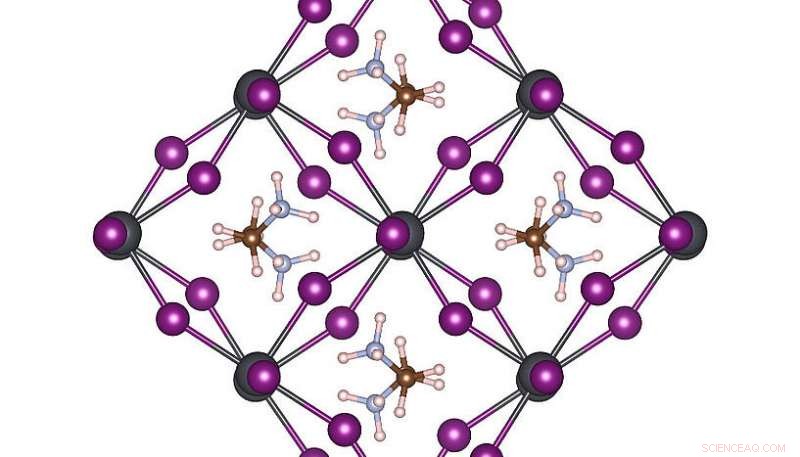
Estrutura atômica de alta simetria de MAPbI3 em temperatura ambiente. Crédito:Menno Bokdam / Universidade de Viena
Na escala atômica, os materiais podem mostrar uma rica paleta de comportamento dinâmico, o que afeta diretamente as propriedades físicas desses materiais. Por muitos anos, tem sido um sonho descrever essa dinâmica em materiais complexos em várias temperaturas usando simulações de computador. Físicos da Universidade de Viena desenvolveram um método de aprendizado de máquina on-the-fly que permite tais cálculos por meio da integração direta com o Vienna Ab-initio Simulation Package (VASP) baseado em mecânica quântica. A versatilidade do método de autoaprendizagem é demonstrada por novas descobertas, publicado no jornal Cartas de revisão física , nas transições de fase de perovskitas híbridas. Essas perovskitas são de grande interesse científico devido ao seu potencial na captação de energia solar e outras aplicações.
Em temperatura ambiente, todos os materiais estão em constante movimento em escala atômica. Mesmo a rocha sólida consiste em átomos que giram. As propriedades físicas dos materiais estão diretamente ligadas ao arranjo dos átomos no, assim chamado, estrutura de cristal. Dependendo da temperatura ou pressão, esse arranjo pode mudar, afetando as propriedades dos materiais. Pode-se pensar em diamante, que é transparente e duro por causa do arranjo periódico de átomos de carbono no cristal de diamante. Os mesmos átomos, arranjado de forma diferente, resulta em preto, grafite quebradiça. Já era possível calcular com precisão as coordenadas dos átomos em materiais simples em diferentes temperaturas com simulações de mecânica quântica de dinâmica molecular (MD). Contudo, tais cálculos são computacionalmente caros e restringem as aplicações práticas a algumas centenas de átomos e tempo de simulação limitado.
Os físicos do grupo de Física de Materiais Computacionais da Universidade de Viena desenvolveram uma nova abordagem que supera essas limitações e torna possíveis as simulações de materiais complexos para futuras aplicações de energia. Isso é alcançado desenvolvendo um algoritmo de autoaprendizagem baseado em dados eficiente e robusto e, mais importante, integrando este algoritmo diretamente no Vienna Ab-initio Simulation Package (VASP). Na nova abordagem, a "máquina" pode pegar, sozinho, os ingredientes essenciais para uma descrição mais simples do modelo dos átomos interagindo durante as simulações de MD. Depois de calcular algumas centenas de intervalos de tempo, a máquina pode prever com precisão suficiente as posições dos átomos em intervalos de tempo consecutivos. A máquina também é capaz de fazer uma estimativa de sua precisão para as etapas consecutivas. Se o erro for muito alto, a máquina muda de marcha e executa o muito preciso, mas caro, Cálculos de MD. Quanto mais o tempo de simulação passa, quanto mais a máquina aprende e mais precisa ela se torna. Desta maneira, cada vez menos cálculos de MD são necessários, o que eventualmente leva à situação em que todos os intervalos de tempo são feitos pela máquina. Além disso, a capacidade de autoaprendizagem instantânea reduz a necessidade de intervenção humana exigida por outros métodos de aprendizagem de máquina existentes.
Para demonstrar o poder deste novo método, os pesquisadores o aplicaram para estudar as transições entre diferentes estruturas atômicas do MAPbI 3 perovskite ao mudar a temperatura. Este material é muito popular devido ao seu potencial como um novo componente barato de célula solar. É feito de moléculas orgânicas que podem girar rapidamente, separados uns dos outros por uma rede composta de átomos de chumbo e iodeto. Dependendo da temperatura, três fases cristalinas diferentes são formadas. Os mecanismos atômicos próximos à temperatura de transição são muito difíceis de serem determinados por experimento, e simulações de MD exigiriam anos de tempo de computação, mesmo em um sistema de supercomputação moderno. Depois de aprender, a máquina pode prever as temperaturas de transição de fase e constantes de rede deste material com precisão sem precedentes. O método desenvolvido é geral e aplicável a muitos outros problemas futuros da ciência dos materiais e estará disponível para pesquisadores de todo o mundo na próxima versão do VASP.
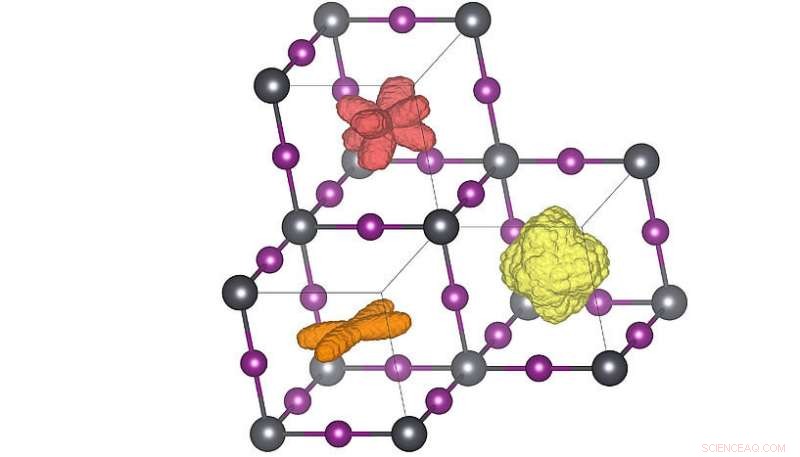
Distribuições tridimensionais da orientação da molécula nas três diferentes fases do cristal. Quando a temperatura é elevada (laranja → vermelho → amarelo) as moléculas podem atingir mais orientações. A distribuição em vermelho corresponde à estrutura da temperatura ambiente. Crédito:Menno Bokdam / Universidade de Viena