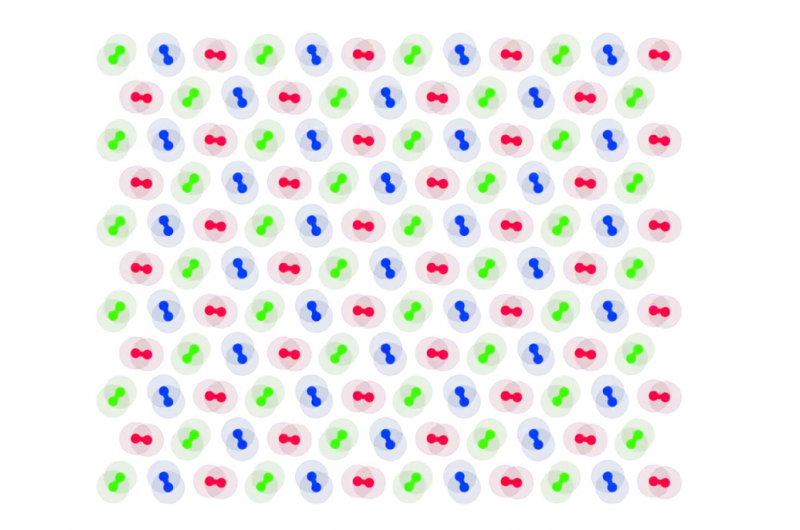
Tensão mecânica, mudanças de pressão ou temperatura ou adição de agentes químicos de dopagem podem levar a uma mudança abrupta de isolador para condutor em materiais como óxido de níquel (ilustrado aqui). Íons de níquel (azul) e íons de oxigênio (vermelho) circundam um íon dopante de potássio (amarelo). Os métodos Quantum Monte Carlo podem prever com precisão as regiões onde a densidade de carga (roxo) se acumulará nesses materiais. Crédito:Anouar Benali, Laboratório Nacional de Argonne
Resolver um problema complexo rapidamente requer trocas cuidadosas - e simular o comportamento dos materiais não é exceção. Para obter respostas que prevejam o funcionamento molecular de forma viável, os cientistas devem trocar aproximações matemáticas que aceleram a computação em detrimento da precisão.
Mas magnetismo, a condutividade elétrica e outras propriedades podem ser bastante delicadas, diz Paul R.C. Kent, do Laboratório Nacional de Oak Ridge do Departamento de Energia (DOE). Essas propriedades dependem da mecânica quântica, os movimentos e interações de uma miríade de elétrons e átomos que formam os materiais e determinam suas propriedades. Os pesquisadores que estudam essas características devem modelar grandes grupos de átomos e moléculas, em vez de apenas alguns. A complexidade deste problema exige o aumento da eficiência e precisão das ferramentas computacionais.
É aí que entra um método chamado modelagem quântica de Monte Carlo (QMC). Muitas outras técnicas aproximam o comportamento dos elétrons como uma média geral, por exemplo, em vez de considerá-los individualmente. QMC permite contabilizar o comportamento individual de todos os elétrons sem grandes aproximações, reduzindo erros sistemáticos em simulações e produzindo resultados confiáveis, Kent diz.
O interesse de Kent no QMC remonta ao seu doutorado. pesquisa na Universidade de Cambridge na década de 1990. No ORNL, ele recentemente voltou ao método porque os avanços tanto no hardware do supercomputador quanto nos algoritmos permitiram aos pesquisadores melhorar sua precisão.
"Podemos fazer novos materiais e uma fração maior de elementos na tabela periódica, "Kent diz." Mais importante, podemos começar a fazer alguns dos materiais e propriedades onde os métodos mais aproximados que usamos no dia a dia não são confiáveis. "
Mesmo com esses avanços, simulações desses tipos de materiais, aqueles que incluem até algumas centenas de átomos e milhares de elétrons, requer levantamento de peso computacional. Kent lidera um Centro de Ciências de Energia Básica do DOE, o Centro de Simulações Preditivas de Materiais Funcionais (CPSFM), que inclui pesquisadores de ORNL, Laboratório Nacional de Argonne, Sandia National Laboratories, Laboratório Nacional Lawrence Livermore, a Universidade da Califórnia, Berkeley e North Carolina State University.
Seu trabalho é apoiado por um impacto computacional inovador e inovador do DOE em teoria e experimentos (INCITE) alocação de 140 milhões de horas de processador, dividida entre os supercomputadores Titan da Oak Ridge Leadership Computing Facility e os supercomputadores Mira da Argonne Leadership Computing Facility. Ambos os centros de computação são instalações do usuário do DOE Office of Science.
Para levar QMC para o próximo nível, Kent e colegas começam com materiais como o dióxido de vanádio, que exibe um comportamento eletrônico incomum. Em temperaturas mais baixas, este material isola contra o fluxo de eletricidade. Mas logo acima da temperatura ambiente, o dióxido de vanádio muda abruptamente sua estrutura e comportamento.
De repente, esse material se torna metálico e conduz eletricidade de forma eficiente. Os cientistas ainda não entendem exatamente como e por que isso ocorre. Fatores como tensão mecânica, a pressão ou dopagem dos materiais com outros elementos também induzem essa rápida transição de isolador para condutor.
Contudo, se cientistas e engenheiros pudessem controlar esse comportamento, esses materiais podem ser usados como interruptores, sensores ou, possivelmente, a base para novos dispositivos eletrônicos. "Essa grande mudança na condutividade de um material é o tipo de coisa que gostaríamos de ser capazes de prever com segurança, "Kent diz.
Pesquisadores de laboratório também estão estudando esses isolantes para condutores com experimentos. Esse esforço de validação confere confiança ao poder preditivo de seus métodos computacionais em uma variedade de materiais. A equipe desenvolveu um software de código aberto, conhecido como QMCPACK, que agora está disponível online e em todos os recursos computacionais do DOE Office of Science.
Kent e seus colegas esperam construir supercondutores de alta temperatura e outros materiais complexos e misteriosos. Embora os cientistas conheçam as amplas propriedades desses materiais, Kent diz, "ainda não podemos relacioná-los à estrutura real e aos elementos dos materiais. Portanto, esse é um grande desafio para o campo da física da matéria condensada."
Os métodos de modelagem da mecânica quântica mais precisos restringem os cientistas a examinar apenas alguns átomos ou moléculas. Quando os cientistas querem estudar sistemas maiores, os custos de computação tornam-se rapidamente pesados. QMC oferece um compromisso:o tamanho de um cálculo aumenta cubicamente em relação ao número de elétrons, um desafio mais gerenciável. O QMC incorpora apenas algumas aproximações controladas e pode ser aplicado aos numerosos átomos e elétrons necessários. É bem adequado para os supercomputadores petaescala de hoje - capazes de cálculos de um quatrilhão ou mais a cada segundo - e os supercomputadores exascale de amanhã, que será pelo menos mil vezes mais rápido. O método mapeia elementos de simulação com relativa facilidade nos nós de computação desses sistemas.
A equipe CPSFM continua a otimizar QMCPACK para supercomputadores cada vez mais rápidos, incluindo a Cúpula do OLCF, que estará totalmente operacional em janeiro de 2019. A maior capacidade de memória nas GPUs Nvidia Volta daquela máquina - 16 gigabytes por unidade de processamento gráfico em comparação com 6 gigabytes no Titan - já aumenta a velocidade de computação. Com a ajuda de Ed D'Azevedo e Andreas Tillack da OLCF, os pesquisadores implementaram algoritmos aprimorados que podem dobrar a velocidade de seus cálculos maiores.
QMCPACK é parte do Projeto de Computação Exascale do DOE, e a equipe já está antecipando desafios de dimensionamento adicionais para executar QMCPACK em máquinas futuras. Para realizar as simulações desejadas em aproximadamente 12 horas em um supercomputador exascale, Kent estima que eles precisarão de algoritmos 30 vezes mais escaláveis do que os da versão atual.
Mesmo com hardware e algoritmos aprimorados, Os cálculos do QMC sempre serão caros. Portanto, Kent e sua equipe gostariam de usar o QMCPACK para entender onde os métodos mais baratos dão errado para que possam melhorá-los. Em seguida, eles podem salvar cálculos QMC para os problemas mais desafiadores na ciência dos materiais, Kent diz. "Idealmente, aprenderemos o que está fazendo com que esses materiais sejam muito difíceis de modelar e, em seguida, melhoraremos as abordagens mais baratas para que possamos fazer varreduras muito mais amplas de diferentes materiais."
A combinação de métodos QMC aprimorados e um conjunto de abordagens de modelagem computacionalmente mais baratas pode abrir caminho para novos materiais e uma compreensão de suas propriedades. Projetar e testar novos compostos em laboratório é caro, Kent diz. Os cientistas poderiam economizar tempo e recursos valiosos se pudessem primeiro prever o comportamento de novos materiais em uma simulação.
Mais, ele observa, métodos computacionais confiáveis podem ajudar os cientistas a entender propriedades e processos que dependem de átomos individuais que são extremamente difíceis de observar usando experimentos. "Esse é um lugar onde há muito interesse em ir atrás da ciência fundamental, previsão de novos materiais e viabilização de aplicações tecnológicas. "