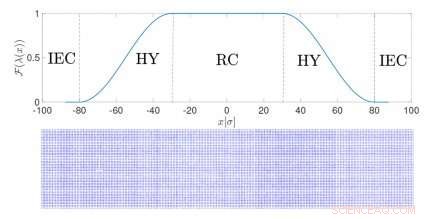
Configuração de uma simulação de resolução adaptativa para sólidos. Crédito:Springer
Simulações de computador são usadas para entender as propriedades da matéria mole, como líquidos, polímeros e biomoléculas como o DNA - que são complicados demais para serem descritos por equações. Muitas vezes são muito caros para simular por completo, dada a intensa potência computacional necessária. Em vez de, uma estratégia útil é acoplar um modelo preciso - aplicado nas áreas do sistema que requerem maior atenção - com um modelo mais simples, modelo idealizado.
Em um artigo recente publicado em EPJ E , Maziar Heidari, do Instituto Max Planck para Pesquisa de Polímeros, Mainz, Alemanha e colegas fazem o modelo preciso em alta resolução coincidir perfeitamente com uma representação exatamente solucionável em resolução mais baixa.
O ideal, modelo mais simples é um tipo de representação nua de átomos ou moléculas, que não interagem entre si. Estudos anteriores aplicaram esta estratégia a líquidos, mas neste estudo, os autores o aplicam pela primeira vez a um sólido modelo acoplado a um cristal ideal, em que os átomos têm movimentos restritos e não interagem, apelidado de cristal de Einstein. A equipe foi capaz de calcular suas propriedades termodinâmicas - por exemplo, temperatura e energia livre - a um custo computacional reduzido.
Neste tipo de simulação, chamadas de simulações de resolução adaptativa, a resolução de uma molécula depende de sua posição no espaço. Na região de transição entre as duas resoluções, as moléculas se adaptam a um modelo ou outro. Esta é uma maneira eficiente de calcular as características termodinâmicas relevantes do sólido real, decompondo-as em uma contribuição ideal - do modelo simplificado - e outro termo, específico para o sistema particular. A metodologia combina a simplicidade dos modelos ideais com a precisão química de representações realistas.