Cientistas computacionais geram conjuntos de dados moleculares em escala extrema
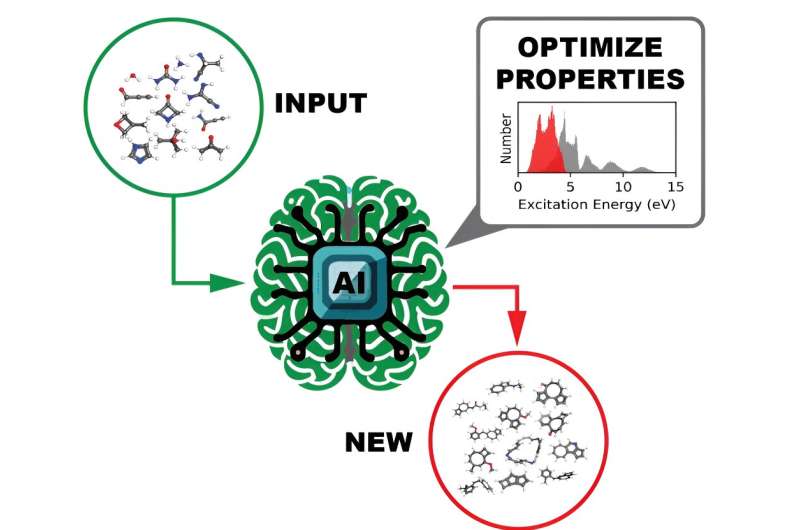
O agente de IA, incorporando um gerador molecular baseado em modelo de linguagem e um preditor de propriedades moleculares baseado em rede neural gráfica, processa um conjunto de moléculas fornecidas pelo usuário (verde) e produz/sugere novas moléculas (vermelho) com a substância química desejada/ propriedades físicas (ou seja, energia de excitação). Crédito:Pilsun Yoo, Jason Smith/ORNL, US DOE Uma equipe de cientistas computacionais do Laboratório Nacional Oak Ridge do Departamento de Energia gerou e divulgou conjuntos de dados em escala sem precedentes que fornecem as propriedades espectrais visíveis no ultravioleta de mais de 10 milhões de moléculas orgânicas. Compreender como uma molécula interage com a luz é essencial para descobrir suas propriedades eletrônicas e ópticas, que por sua vez têm aplicações fotoativas potenciais em produtos como células solares ou sistemas de imagens médicas.
Usando recursos de computação de alto desempenho no Oak Ridge Leadership Computing Facility, a equipe do ORNL executou cálculos de química quântica para gerar vastos conjuntos de dados. Para cada uma dessas moléculas orgânicas, a equipe executou cálculos de modelagem de material atomístico com várias aproximações para calcular diferentes propriedades de interesse no estado excitado. As descobertas da equipe foram publicadas em Scientific Data .
O uso final pretendido para os conjuntos de dados de código aberto é treinar um modelo de aprendizagem profunda para identificar moléculas com propriedades optoeletrônicas e de fotorreatividade personalizadas, uma abordagem que é muito mais rápida e fácil de conduzir do que os métodos atuais.
“O uso de modelos DL para design molecular é essencial porque o espaço químico que deve ser explorado para a busca dessas moléculas é extremamente grande”, disse o autor principal Massimiliano Lupo Pasini, cientista de dados da Divisão de Ciências Computacionais e Engenharia do ORNL.
"Tanto os experimentos quanto os cálculos de primeiros princípios existentes, que são baseados nas leis físicas que determinam como a matéria e a energia interagem no nível subatômico, são simplesmente inacessíveis por diferentes razões. Os experimentos exigem muito trabalho e os cálculos de primeiros princípios podem facilmente prejudicar a supercomputação Mas os modelos DL fornecem ferramentas muito promissoras para superar essas barreiras", disse Lupo Pasini.
O projeto começou quando Stephan Irle, líder do grupo de Química Computacional e Ciências de Nanomateriais do ORNL, identificou os espectros ultravioleta-visíveis das moléculas como uma propriedade útil para prever com modelos DL.
Construir um modelo DL suficientemente complexo para identificar propriedades moleculares desejáveis requer treiná-lo com enormes volumes de dados que exploram todas as diferentes regiões do espaço químico. Quanto mais dados forem coletados, mais o modelo DL treinado neles poderá alcançar a robustez e generalização necessárias para funcionar de maneira eficaz. No entanto, a coleta de volumes tão grandes de dados científicos para DL escalonável pode apresentar problemas de fluxo de dados, especialmente em instalações com múltiplos usuários, como o OLCF, uma instalação de usuários do DOE Office of Science localizada no ORNL.
“Um desafio que ocorre ao gerar grandes volumes de dados é que o número de arquivos a serem gerenciados aumenta drasticamente. Se não for gerenciado corretamente, um volume tão grande de dados pode comprometer o funcionamento do sistema de arquivos paralelo, que é um componente importante do estado. instalações HPC de última geração", disse Lupo Pasini.
Para enfrentar esse desafio, Lupo Pasini colaborou com o cientista da computação do ORNL, Kshitij Mehta, para desenvolver um software de fluxo de trabalho escalável que garanta que os arquivos gerados pelo código da mecânica quântica sejam tratados adequadamente sem sobrecarregar o sistema de arquivos, como o Orion do OLCF, que é um arquivo compartilhado recurso que lida com a entrada, saída e armazenamento de dados em sistemas de supercomputadores.
Como teste de prova de conceito, a equipe gerou o conjunto de dados GDB-9-Ex de 96.766 moléculas compostas de carbono, nitrogênio, oxigênio e flúor, com no máximo nove átomos diferentes de hidrogênio. Mostrou que o fluxo de trabalho projetado é eficaz e que o treinamento DL prevê com precisão a posição e a intensidade dos picos mais relevantes do espectro ultravioleta-visível.
A partir desse sucesso inicial, a equipe aumentou seu volume com o conjunto de dados ORNL_AISD-Ex, que contém 10.502.917 moléculas compostas de carbono, nitrogênio, oxigênio, flúor e enxofre, com no máximo 71 átomos diferentes de hidrogênio. Pilsun Yoo, pesquisador de pós-doutorado do grupo de Irle, desenvolveu ferramentas para analisar os conjuntos de dados resultantes.
O espectro ultravioleta-visível, que descreve os modos de excitação de uma molécula, foi calculado para cada uma das mais de 10 milhões de moléculas. Esta informação revela qual frequência de luz é necessária para atingir uma molécula e quebrar algumas ligações do composto químico.
Outra propriedade de interesse calculada para cada molécula foi a lacuna HOMO-LUMO – a lacuna de energia entre o orbital molecular mais ocupado e o orbital molecular desocupado mais baixo – que mede de forma confiável a estabilidade da molécula. Com essas informações, um modelo DL poderia filtrar com eficiência os dados para identificar moléculas promissoras para diferentes usos prospectivos.
Na verdade, Lupo Pasini e sua equipe no ORNL, incluindo o cientista computacional em aprendizado de máquina Pei Zhang e o cientista de pesquisa de dados de HPC Jong Youl Choi, estão desenvolvendo exatamente esse modelo DL:HydraGNN.
"A arquitetura HydraGNN pega a estrutura atômica, converte-a em um gráfico e então tenta prever como saída o que o código de primeiros princípios produziria. É um modelo substituto para cálculos caros de primeiros princípios", disse Lupo Pasini.
Os resultados do treinamento da HydraGNN nos conjuntos de dados e suas descobertas moleculares serão detalhados em um próximo artigo.
Mais informações: Massimiliano Lupo Pasini et al, Dois conjuntos de dados de estado excitado para espectros UV-vis químicos quânticos de moléculas orgânicas, Scientific Data (2023). DOI:10.1038/s41597-023-02408-4 Informações do diário: Dados científicos