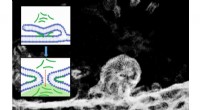
Estruturas geradas e redes de caminhos de reação para as etapas de reação previstas para a reação de Strecker (a) e reação de Passerini (b). As setas brancas mostram o caminho de várias etapas correspondente ao mecanismo de reação conhecido. Crédito:Satoshi Maeda
Os pesquisadores superaram as limitações computacionais para prever os materiais de partida de reações em várias etapas usando apenas informações sobre a molécula do produto alvo. Seu estudo está publicado em
JACS Au .
Você já pegou o final de um programa de TV e se perguntou como a história progrediu para esse final? De maneira semelhante, os químicos geralmente têm uma molécula desejada em mente e se perguntam que tipo de reação poderia produzi-la. Pesquisadores do Grupo Maeda do Institute for Chemical Reaction Design and Discovery (ICReDD) e da Hokkaido University desenvolveram um método que pode prever a "história" (ou seja, os materiais de partida e os caminhos de reação) de reações químicas de várias etapas usando apenas informações sobre a "terminação" (isto é, as moléculas do produto).
Prever a receita de uma molécula de produto alvo, sem outro conhecimento além da própria molécula, seria uma ferramenta poderosa para acelerar a descoberta de novas reações. O grupo Maeda desenvolveu anteriormente um método computacional que conseguiu prever reações de etapa única dessa maneira. No entanto, a expansão para reações de várias etapas leva a um aumento dramático no número de possíveis vias de reação - o que é conhecido como explosão combinatória. Esse aumento acentuado na complexidade resulta em custos de cálculo proibitivamente altos.
Para superar essa limitação, os pesquisadores desenvolveram um algoritmo que reduz o número de caminhos que precisam ser explorados, descartando caminhos menos viáveis em cada etapa da reação. Depois de calcular todos os caminhos possíveis para um passo para trás na reação, um método de análise cinética avalia quão bem cada caminho produz a molécula alvo. Os caminhos de reação que não produzem a molécula alvo acima de uma porcentagem de limite pré-definida são considerados não significativos o suficiente e não são explorados mais.
Este ciclo de explorar, avaliar e descartar caminhos de reação é repetido para cada passo para trás em uma reação de várias etapas e mitiga a explosão combinatória que normalmente ocorreria, tornando as reações de várias etapas mais viáveis para calcular. Os métodos anteriores eram limitados a reações de etapa única, enquanto esse novo método era capaz de prever reações que envolviam mais de seis etapas, marcando um grande salto na capacidade.
Como um teste de prova de conceito, os pesquisadores testaram o método em duas reações de várias etapas bem conhecidas, as reações de Strecker e Passerini. Milhares de candidatos a material de partida foram propostos para cada reação, que foram filtrados para os candidatos mais promissores com base na estabilidade e rendimento do produto. Criticamente, entre os candidatos propostos estavam os materiais de partida bem conhecidos para cada reação, confirmando a capacidade da técnica de identificar materiais de partida experimentalmente viáveis apenas a partir da molécula do produto alvo.
Embora seja necessário mais trabalho para permitir a previsão de sistemas ainda maiores e mais complexos, os pesquisadores antecipam que esse avanço no manuseio de processos de várias etapas acelerará a descoberta de novas reações químicas.
“Este trabalho fornece uma abordagem única, pois é a primeira vez que é possível realizar previsões reversas de reações de várias etapas usando cálculos químicos quânticos sem usar nenhum conhecimento ou dados sobre a reação”, disse o professor Satoshi Maeda. “Esperamos que esta técnica permita a descoberta de transformações químicas totalmente inimagináveis, caso em que há pouco conhecimento ou dados experimentais para usar”.
+ Explorar mais Novo método de síntese química assistida por computador reduz tempo e custo de pesquisa