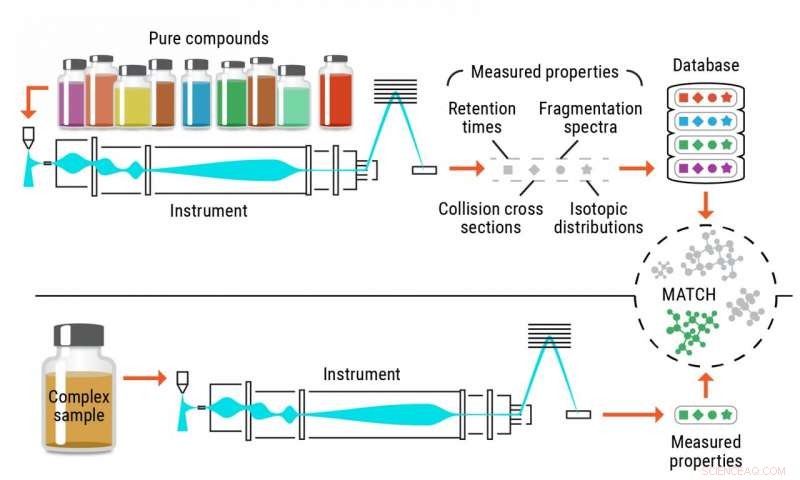
 p Ilustração do processo convencional de identificação de metabólitos. Crédito:Pacific Northwest National Laboratory
p Ilustração do processo convencional de identificação de metabólitos. Crédito:Pacific Northwest National Laboratory
p Identificação precisa de metabólitos, e outros pequenos produtos químicos, em amostras biológicas e ambientais historicamente ficou aquém do uso de métodos tradicionais. As táticas convencionais dependem de compostos de referência puros, chamados padrões, para reconhecer as mesmas moléculas em amostras complexas. Essas abordagens são limitadas pela disponibilidade de produtos químicos puros que são usados como padrões. p "Queríamos realmente contornar o paradigma atual de como um experimento de metabolômica é conduzido e como as moléculas são identificadas com segurança, "disse Tom Metz, cientista biomédico do Pacific Northwest National Laboratory (PNNL) e diretor do Pacific Northwest Advanced Compound Identification Core.
p Um problema com o método atual é que existem poucos compostos puros que os pesquisadores podem comprar dos fornecedores; a maioria dos fornecedores tem acesso a cerca de 3, 000–4, 000 compostos.
p "Se você considerar o que está previsto que ocorra na natureza, você está olhando para> 1030 compostos ou mais que poderiam ser possíveis, "disse Metz." Então, quando você compara os poucos milhares de produtos químicos padrão aos quais você tem acesso com o grande número de compostos potenciais, você não está nem perto. "
p
Abordagem de identificação livre de padrões
p Para resolver este problema, Metz e sua equipe no PNNL conceituaram uma abordagem - metabolômica sem padrões - com a qual eles calculam ou prevêem informações sobre propriedades múltiplas para moléculas de interesse, a fim de gerar bibliotecas de referência abrangentes e, em seguida, combinar dados experimentais contendo as mesmas propriedades para essas bibliotecas, permitindo a identificação do composto.
p Usando essa nova abordagem, pesquisadores enviam estruturas químicas por meio de programas de aprendizado de máquina ou química quântica para prever com precisão as propriedades experimentais dos metabólitos.
p "Se formos precisos o suficiente nessas previsões, teoricamente nunca precisaríamos analisar um composto puro novamente, "disse Metz." Esta coleção de ferramentas mudará o paradigma atual em metabolômica, e em um futuro próximo haverá algumas aplicações realmente boas para mostrar à comunidade de pesquisa os benefícios desta nova abordagem. "
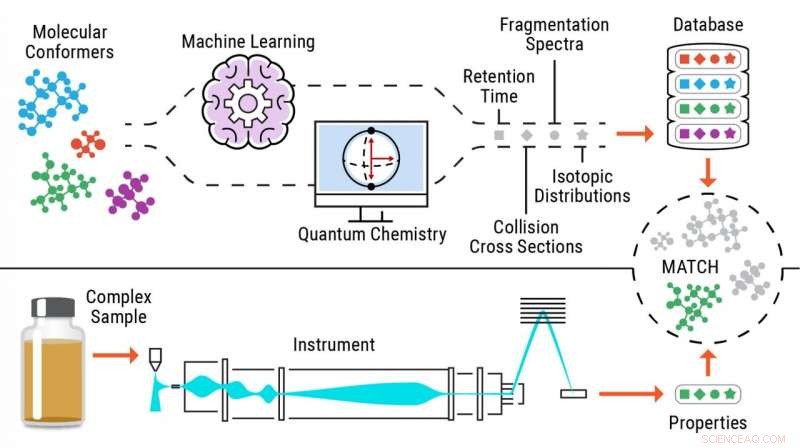 p Ilustração do processo de identificação livre de padrões de metabólitos. Crédito:Pacific Northwest National Laboratory
p Ilustração do processo de identificação livre de padrões de metabólitos. Crédito:Pacific Northwest National Laboratory
p Por não ter que confiar em dados de análises de padrões puros para identificar pequenas moléculas, a abordagem livre de padrões permite a identificação de até 90 por cento mais produtos químicos em amostras e torna essas ferramentas computacionais altamente úteis em várias áreas de aplicação, incluindo a descoberta de novas drogas, química forense, e pesquisa ambiental e biomédica.
p "Por exemplo, no projeto de um novo medicamento, o usuário seria capaz de dizer:'Eu tenho um certo número de propriedades com essas drogas certas, mas eles são tóxicos. Podemos prever um composto que teria propriedades semelhantes, mas pode não ser tóxico? '", Disse Metz." Se os dados de treinamento corretos pudessem ser fornecidos ao programa DarkChem, DarkChem poderia então realizar essa previsão. "
p
Pacote personalizável de programas
p A nova abordagem para identificação metabolômica livre de padrões usa quatro ferramentas-chave para gerar, em bibliotecas de referência de metabólitos derivados de silico, e extrair e combinar dados experimentais para produzir identificações de compostos:
- In Silico Chemical Library Engine (ISiCLE), um computador de alto desempenho amigável, abordagem da química quântica para gerar propriedades químicas previstas.
- DarkChem, um autoencoder variacional que aprende uma representação numérica ou latente contínua da estrutura molecular, que pode caracterizar e expandir bibliotecas de referência.
- Extração de dados para espectrometria multidimensional integrada (DEIMoS), uma ferramenta de software modular que pode extrair recursos de dados coletados em plataformas analíticas multidimensionais.
- Mecanismo de correspondência de atributos múltiplos (MAME), que combina dados experimentais com bibliotecas de referência com base em vários atributos químicos.
p As ferramentas foram projetadas para funcionar juntas, mas também podem ser usados separadamente. Os pesquisadores podem personalizar os diferentes aplicativos com base nas necessidades de um cliente ou áreas de pesquisa, criando uma abordagem totalmente modular.
p
Avançando em um campo de pesquisa
p Agora mesmo, na comunidade metabolômica, todos os pesquisadores identificam o mesmo conjunto de moléculas em cada amostra. A razão para isso é que todos eles têm os mesmos compostos puros que compraram para construir suas bibliotecas de referência.
p "Nossa visão é que, ao usar a abordagem livre de padrões, você nunca será limitado pela extensão de pequenas moléculas que podem ser identificadas em uma amostra, "disse Metz." Isso é realmente uma virada de jogo para a metabolômica. E é muito emocionante ver o que o próximo ano ou mais nos reserva para isso. "