Crédito:Universidade de Leiden
As cadeias de proteínas normalmente dobram para funcionar. O dobramento é um processo complexo e, se feito corretamente, leva a uma topologia de dobra funcional única para uma determinada cadeia de proteína. Outras topologias também são possíveis, mas geralmente não são funcionais ou são tóxicas. Essas proteínas mal dobradas são então desdobradas e subsequentemente redobradas para a topologia de dobra correta; de outra forma, eles sofrem degradação.
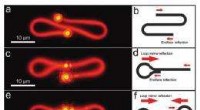
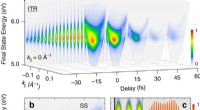
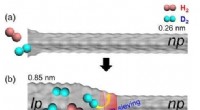
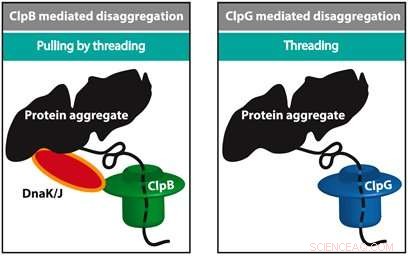
Várias máquinas, incluindo ClpB e ClpG, são responsáveis por desdobrar uma proteína dobrada. O ClpB trabalha em estreita colaboração com o HSP70 (DnaK) e o HSP40 (DnaJ) e usa energia para desdobrar uma cadeia, enquanto o ClpG não depende do HSP70. Uma grande questão é por que as células são equipadas com diferentes tipos de máquinas e o que determina a eficiência do desdobramento. Alireza Mashaghi e sua equipe no LACDR / Universidade de Leiden resolveram esse quebra-cabeça monitorando o desdobramento de modelos de cadeia mal dobrados no nível de uma única molécula. Três abordagens de desdobramento foram comparadas, nomeadamente, passando por um poro, puxando pelas pontas, e puxando por rosca.
Os resultados desta análise, que são publicados em 25 de outubro no Journal of Physical Chemistry B , revelam que a topologia do circuito da cadeia dobrada determina criticamente o número de caminhos e a eficiência do desdobramento de uma maneira que depende da abordagem mecânica empregada. O estudo fornece informações sobre os mecanismos de desdobramento da proteína celular. Esses achados podem ajudar na seleção de alvos ideais de acompanhantes para farmacoterapia de doenças de dobramento incorreto.