Figura 1:ângulo diedro (o ângulo formado pelo plano criado pelos átomos A, B, e C, e o plano criado pelos átomos B, C, e D). Crédito:Fujitsu
Fujitsu Laboratories anunciou hoje o desenvolvimento de tecnologia de simulação molecular para a descoberta de drogas que podem estimar com precisão a afinidade de ligação, que representa o grau em que proteínas que podem causar doenças (proteínas-alvo) se ligam a substâncias químicas que poderiam se tornar drogas candidatas. No processo de descoberta de drogas, há uma demanda por uma previsão precisa da afinidade de ligação entre as proteínas alvo e as substâncias químicas, que oferece uma estimativa aproximada da eficácia de um medicamento. A tecnologia de simulação molecular foi amplamente usada no passado como um método de previsão da afinidade de ligação, calcular as forças aproximadas que surgem entre os átomos nas moléculas usando a mecânica newtoniana. O problema com este método, Contudo, permanece o baixo grau de precisão de sua estimativa dos parâmetros mais importantes - o grau de torção nos locais de ligação. Isso significa que a precisão de sua estimativa da afinidade de ligação geral também é pobre.
Agora, Fujitsu Laboratories desenvolveu tecnologia de simulação molecular que estima o grau de torção em uma substância química, que está diretamente conectado à afinidade de ligação prevista. A nova tecnologia não leva em consideração apenas o local de colagem onde ocorrerá a torção, mas também o impacto dos átomos vizinhos. Fujitsu Laboratories avaliou esta tecnologia para 190 tipos de substâncias químicas, comparar os resultados com os resultados corretos obtidos a partir do cálculo dos primeiros princípios e, em seguida, avaliar a taxa de erro. Ao fazer isso, foi capaz de confirmar que a taxa de erro na estimativa do grau de torção foi, na média, um décimo da tecnologia anterior. Prevê-se que o uso desta nova tecnologia na descoberta de medicamentos baseados em TI, com sua capacidade de estimar com precisão a afinidade de ligação de proteínas alvo e substâncias químicas, oferece o potencial para esforços inovadores de descoberta de novos medicamentos que não poderiam ser alcançados com abordagens anteriores.
A descoberta de novos medicamentos requer despesas e prazos significativos que podem ser medidos em décadas, levando a uma busca global por novos métodos de descoberta de drogas. Um dos métodos que tem recebido considerável interesse é a descoberta de medicamentos baseados em TI, um novo método de descoberta de drogas por meio de computadores que possibilita a criação de substâncias químicas como candidatas a novas drogas com alta probabilidade de sucesso. A descoberta de medicamentos com base em TI tornou-se um ponto focal para as expectativas como uma tecnologia inovadora para a criação de novos medicamentos, porque, ao contrário dos métodos anteriores de tentativa e erro, em que as substâncias químicas são criadas e testadas repetidamente, esta abordagem torna possível projetar virtualmente substâncias químicas e estimar seus efeitos.
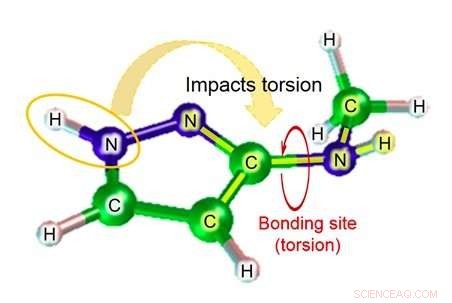
Figura 2:Exemplo de estrutura molecular:3- (metilamino) pirazol. Crédito:Fujitsu
Os efeitos de uma substância química como medicamento são expressos quando a substância química se liga a uma proteína-alvo. Quando a substância química se liga à proteína alvo, ele pode mudar sua forma de acordo com a da proteína-alvo. O grau de deformação, nomeadamente, os parâmetros que indicam a extensão dessa mudança de forma, está diretamente ligado à afinidade de ligação da substância e da proteína, e dá uma ideia aproximada de seu efeito como droga. Diante disso, há uma forte demanda pela capacidade de prever esse valor com precisão. Para calcular o grau de deformação de uma substância química, existem métodos baseados na mecânica quântica e métodos baseados na mecânica newtoniana. O cálculo dos primeiros princípios com base na mecânica quântica permite cálculos extremamente precisos, resolver os estados dos elétrons a partir dos tipos e posições dos átomos envolvidos. Por outro lado, Contudo, a capacidade dos primeiros princípios de realizar cálculos exatos leva necessariamente a um tempo massivo necessário para completar os cálculos. A fim de simular o grau de deformação de inúmeras substâncias químicas, o tempo necessário é da ordem de anos, tornando este método impraticável. Por outro lado, cálculos aproximados baseados em simulações moleculares são extremamente rápidos, usando a mecânica newtoniana para calcular as forças entre os átomos dentro das moléculas, e pode até mesmo lidar com moléculas grandes como proteínas com bastante facilidade. Consequentemente, este método é amplamente utilizado. Com a mecânica newtoniana, as forças entre os átomos são expressas da seguinte maneira:
Entre estes, quando uma substância química é ligada a uma proteína alvo, o grau de torção da ligação representa o grau importante de deformação. Com a tecnologia existente, Contudo, a precisão da estimativa do parâmetro do ângulo diedro (figura 1), que é necessário para calcular o grau de torção da ligação, é bastante baixo, resultando no problema de baixa precisão na estimativa da afinidade do título na simulação.
A Fujitsu Laboratories vem desenvolvendo tecnologia de simulação molecular há mais de dez anos. Agora, usando o conhecimento obtido por meio de esforços anteriores, A Fujitsu Laboratories desenvolveu uma tecnologia de simulação molecular que pode estimar o parâmetro do ângulo diédrico levando em consideração o impacto dos átomos próximos à ligação. A tecnologia existente estima o parâmetro do ângulo diédrico com base em um total de quatro átomos - os dois átomos na ligação relevante, e os outros átomos aos quais cada um desses átomos estava ligado. Dependendo da estrutura da molécula, Contudo, há casos em que átomos além desses quatro podem ter um impacto significativo, e nesses casos, a margem de erro da estimativa pode ser bastante grande. Com esta tecnologia, A Fujitsu Laboratories criou um banco de dados de fórmulas de estimativa para padrões de estrutura parcial, onde o impacto de átomos mais distantes do local do vínculo pode ser significativo, bem como para o grau de torção de substâncias químicas que seria esperado nesse caso. Usando a fórmula de estimativa relevante para encontrar o grau de torção (figura 2) no caso de moléculas correspondentes ao banco de dados para estruturas parciais, tornou-se possível até mesmo fazer estimativas altamente precisas para torção molecular, o que antes era difícil de calcular com precisão.
Quando a Fujitsu Laboratories integrou esta tecnologia ao software que havia desenvolvido para gerar parâmetros sofisticados para as forças entre os átomos (FF-FOM), foi capaz de confirmar que os resultados estavam de acordo com cálculos precisos.

Figura 3:Avaliação do desempenho dos valores dos parâmetros do ângulo diédrico usando 190 tipos de estruturas de compostos químicos. Crédito:Fujitsu
Quando a Fujitsu Laboratories avaliou a diferença entre os resultados desta tecnologia e os resultados de um cálculo dos primeiros princípios para a estimativa do grau de torção com 190 tipos de substâncias químicas, era menos de um décimo da tecnologia anterior, na média, 0,6 kcal / mol abaixo das flutuações térmicas da temperatura ambiente, confirmando que a nova tecnologia é prática. Porque pode estimar com precisão a afinidade de ligação de proteínas alvo e substâncias químicas, espera-se que o uso dessa tecnologia leve à criação de novos medicamentos inovadores por meio de seu uso na descoberta de medicamentos baseados em TI.