 p As redes neurais permitem simulações precisas na ciência dos materiais - até o nível de átomos individuais. Crédito:Pascal Friederich, KIT
p As redes neurais permitem simulações precisas na ciência dos materiais - até o nível de átomos individuais. Crédito:Pascal Friederich, KIT
p Pesquisar, desenvolvimento, e a produção de novos materiais depende muito da disponibilidade de métodos de simulação rápidos e ao mesmo tempo precisos. Aprendizado de máquina, em que a inteligência artificial (IA) adquire e aplica autonomamente novos conhecimentos, em breve permitirá aos pesquisadores desenvolver sistemas de materiais complexos em um ambiente puramente virtual. Como é que isso funciona, e quais aplicativos serão beneficiados? Em um artigo publicado no
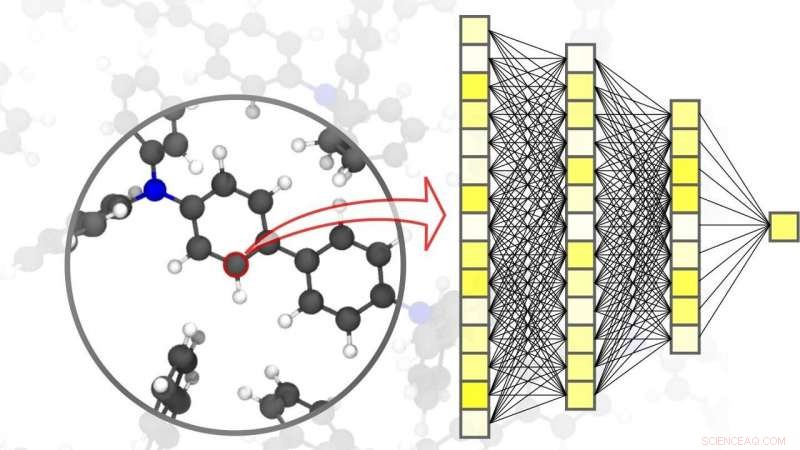
Materiais da Natureza Diário, um pesquisador do Karlsruhe Institute of Technology (KIT) e seus colegas de Göttingen e Toronto explicam tudo. p A digitalização e a virtualização estão se tornando cada vez mais importantes em uma ampla gama de disciplinas científicas. Uma dessas disciplinas é a ciência dos materiais:pesquisa, desenvolvimento, e a produção de novos materiais depende muito da disponibilidade de métodos de simulação rápidos e ao mesmo tempo precisos. Esse, por sua vez, é benéfico para uma ampla gama de diferentes aplicações - desde sistemas eficientes de armazenamento de energia, como os indispensáveis para a utilização de energias renováveis, para novos medicamentos, para cujo desenvolvimento é necessária uma compreensão de processos biológicos complexos. Os métodos de IA e de aprendizado de máquina podem levar as simulações em ciências materiais para o próximo nível. "Comparado aos métodos convencionais de simulação baseados em cálculos de mecânica quântica ou clássica, o uso de redes neurais especificamente adaptadas para simulações de materiais nos permite obter uma vantagem significativa de velocidade, "explica o físico e especialista em IA, Professor Pascal Friederich, Chefe do grupo de pesquisa AiMat — Inteligência Artificial para Ciências dos Materiais no Instituto de Informática Teórica do KIT (ITI). "Com sistemas de simulação mais rápidos, os cientistas serão capazes de desenvolver sistemas de materiais maiores e mais complexos em um ambiente puramente virtual, e entendê-los e otimizá-los até o nível atômico. "
p
Alta precisão do átomo ao material
p Em um artigo publicado em
Materiais da Natureza , Pascal Friederich, que também é líder de grupo associado da divisão Nanomaterials by Information-Guided Design no Instituto de Nanotecnologia do KIT (INT), presentes, juntamente com pesquisadores da Universidade de Göttingen e da Universidade de Toronto, uma visão geral dos princípios básicos do aprendizado de máquina usados para simulações em ciências dos materiais. Isso também inclui o processo de aquisição de dados e métodos de aprendizagem ativa. Os algoritmos de aprendizado de máquina não só permitem que a inteligência artificial processe os dados de entrada, mas também para encontrar padrões e correlações em grandes conjuntos de dados, Aprenda com eles, e fazer previsões e decisões autônomas. Para simulações em ciência dos materiais, é importante alcançar alta precisão em diferentes escalas de tempo e tamanho, variando do átomo ao material, enquanto limita os custos computacionais. Em seu artigo, os cientistas também discutem várias aplicações atuais, como pequenas moléculas orgânicas e grandes biomoléculas, sólido estruturalmente desordenado, líquido, e materiais gasosos, bem como sistemas cristalinos complexos - por exemplo, estruturas metal-orgânicas que podem ser usadas para armazenamento de gás ou para separação, para sensores ou para catalisadores.
p
Ainda mais velocidade com métodos híbridos
p Para estender ainda mais as possibilidades de simulações de materiais no futuro, os pesquisadores de Karlsruhe, Göttingen, e Toronto sugere o desenvolvimento de métodos híbridos:estes combinam métodos de aprendizado de máquina (ML) e mecânica molecular (MM). As simulações MM usam os chamados campos de força para calcular as forças que atuam em cada partícula individual e, assim, prever os movimentos. Como os potenciais dos métodos ML e MM são bastante semelhantes, uma integração estreita com áreas de transição variáveis é possível. Esses métodos híbridos podem acelerar significativamente a simulação de grandes biomoléculas ou reações enzimáticas no futuro, por exemplo.