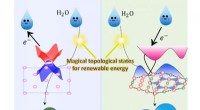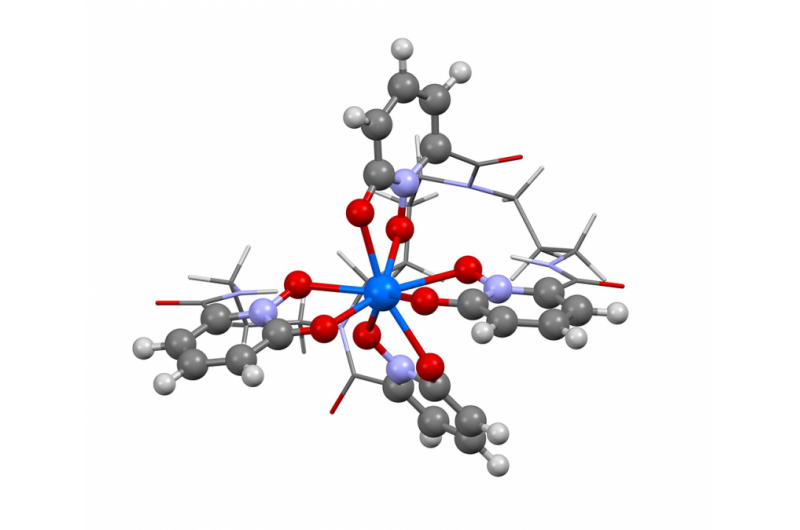
Esta imagem mostra a estrutura do berquélio no estado de oxidação + IV. Os pesquisadores usaram o novo algoritmo do Berkeley Lab para calcular o espectro de absorção e confirmar o que vários resultados experimentais têm sugerido - que o elemento berquélio se quebra com seus pares de elementos pesados, assumindo uma carga positiva extra quando ligado a uma molécula orgânica sintética. Esta propriedade pode ajudar os cientistas a desenvolver métodos melhores para o manuseio e purificação de materiais nucleares. Crédito:Bert de Jong, Berkeley Lab
Objetos que brilham no escuro parecem mágicos quando você é criança - eles podem iluminar um quarto escuro sem a necessidade de eletricidade, pilhas ou uma lâmpada. Então, em algum ponto, você aprende a ciência por trás desse fenômeno. Compostos químicos chamados cromóforos tornam-se energizados, ou animado, quando absorvem a luz visível. Conforme eles voltam ao seu estado normal, a energia armazenada é liberada como luz, que percebemos como um brilho. Na ciência dos materiais, pesquisadores contam com um fenômeno semelhante para estudar as estruturas dos materiais que eventualmente serão usados na catálise química, baterias, aplicações solares e muito mais.
Quando uma molécula absorve um fóton - a partícula fundamental da luz - os elétrons no sistema molecular são promovidos de um estado de baixa energia (fundamental) para um estado de alta energia (excitado). Essas respostas ressoam em frequências de luz específicas, deixando "impressões digitais espectrais" que iluminam as estruturas atômicas e eletrônicas do sistema em estudo.
Em experimentos, as "impressões digitais espectrais" ou espectro de absorção, são medidos com instalações de última geração, como a Advanced Light Source (ALS) no Lawrence Berkeley National Laboratory do Departamento de Energia dos EUA (Berkeley Lab). Em simulações de computador, essas medições são normalmente capturadas com um método de mecânica quântica denominado Teoria Funcional da Densidade Dependente do Tempo (TDDFT). Os modelos computacionais são essenciais para ajudar os pesquisadores a aproveitar ao máximo seus experimentos, prevendo e validando resultados.
No entanto, apesar de sua utilidade, há momentos em que o TDDFT não pode ser usado para calcular o espectro de absorção de um sistema porque exigiria muito tempo e recursos do computador. É aqui que um novo "atalho" matemático desenvolvido por pesquisadores da Divisão de Pesquisa Computacional (CRD) do Berkeley Lab se torna útil. Seu algoritmo acelera os cálculos de absorção por um fator de cinco, portanto, as simulações que costumavam levar de 10 a 15 horas para serem computadas agora podem ser feitas em aproximadamente 2,5 horas.
Um artigo que descreve este método foi publicado no Journal of Chemical Theory and Computation (JCTC). E a nova abordagem para calcular o espectro de absorção será incorporada em um próximo lançamento do amplamente utilizado pacote de software de química computacional NWChem ainda este ano.
Novos algoritmos levam a economia computacional
Para estudar a estrutura química de novas moléculas e materiais, os cientistas normalmente sondam o sistema com um estímulo externo - normalmente um laser - e, em seguida, procuram por pequenas mudanças eletrônicas. Matematicamente, essa mudança eletrônica pode ser expressa como um problema de autovalor. Ao resolver este problema de autovalor, os pesquisadores podem obter uma boa aproximação do espectro de absorção, que por sua vez revela as frequências ressonantes do sistema que está sendo estudado. Enquanto isso, o autovetor correspondente é usado para calcular a intensidade com que o sistema respondeu ao estímulo. Este é essencialmente o princípio por trás da abordagem TDDFT, que foi implementado em vários pacotes de software de química quântica, incluindo o pacote de software de código aberto NWChem.
Embora essa abordagem tenha provado ser bem-sucedida, ele tem limitações para grandes sistemas. Quanto mais ampla a faixa de energia das respostas eletrônicas que um pesquisador tenta capturar em um sistema, quanto mais autovalores e autovetores precisam ser calculados, o que também significa que mais recursos de computação são necessários. Em última análise, o espectro de absorção de um sistema molecular com mais de 100 átomos torna-se proibitivamente caro para computar com este método.
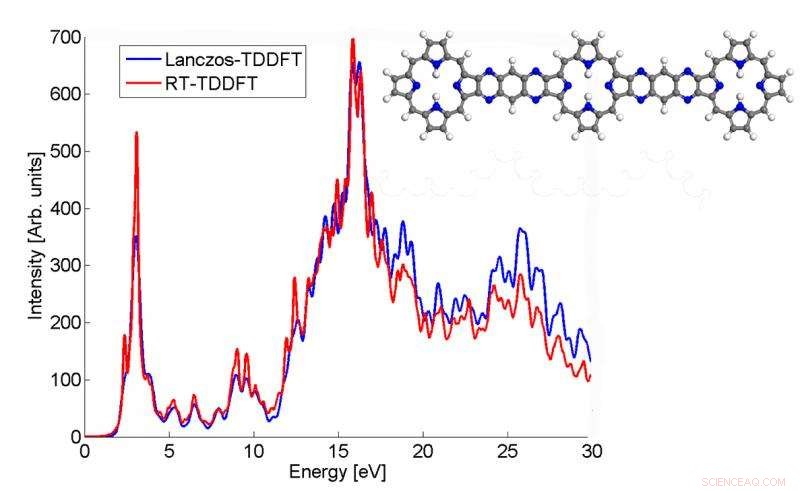
Este gráfico mostra como o espectro de absorção de uma molécula p3b2 calculada pelo algoritmo Lanczos combina com o resultado TDDFT em tempo real. Crédito:Chao Yang, Berkeley Lab
Para superar essas limitações, matemáticos no CRD desenvolveram uma técnica para calcular o espectro de absorção diretamente sem computar explicitamente os autovalores da matriz.
"Tradicionalmente, pesquisadores tiveram que calcular os autovalores e autovetores de matrizes muito grandes para gerar o espectro de absorção, mas percebemos que você não precisa calcular cada valor próprio para obter uma visão precisa do espectro de absorção, "diz Chao Yang, um matemático CRD que liderou o desenvolvimento da nova abordagem.
Ao reformular o problema como uma aproximação de função de matriz, fazendo uso de uma transformação especial e aproveitando a simetria subjacente em relação a uma métrica não euclidiana, Yang e seus colegas foram capazes de aplicar o algoritmo Lanczos e um Método Polinomial de Kernal (KPM) para aproximar o espectro de absorção de várias moléculas. Ambos os algoritmos requerem memória relativamente baixa em comparação com alternativas não simétricas, que é a chave para a economia computacional.
Como este método requer menos poder de computação para atingir um resultado, os pesquisadores também podem calcular facilmente o espectro de absorção para sistemas moleculares com várias centenas de átomos.
"Este método é um passo significativo porque nos permite modelar o espectro de absorção de sistemas moleculares de centenas de átomos com menor custo computacional." diz Niranjan Govind, um químico computacional do Pacific Northwest National Laboratory que colaborou com a equipe do Berkeley Lab no desenvolvimento do método no programa de química computacional NWChem.
Recentemente, cientistas do Berkeley Lab usaram esse método para calcular o espectro de absorção e confirmar o que vários resultados experimentais têm sugerido - que o elemento berquélio se quebra com seus pares de elementos pesados, assumindo uma carga positiva extra quando ligado a uma molécula orgânica sintética. Esta propriedade pode ajudar os cientistas a desenvolver métodos melhores para o manuseio e purificação de materiais nucleares. Um artigo destacando esse resultado foi publicado em 10 de abril na revista. Química da Natureza .
"Os resultados experimentais sugeriam este comportamento incomum no berquélio, mas não havia evidências experimentais suficientes para dizer sim, 100 por cento, isso é o que estamos vendo, "diz o co-autor do estudo Wibe Albert de Jong, um cientista CRD. "Para ter 100 por cento de certeza, fizemos grandes simulações computacionais e as comparamos com os dados experimentais e determinamos que eram, na verdade, vendo berquélio em um estado de oxidação incomum. "
Este novo algoritmo foi desenvolvido por meio de um projeto de Descoberta Científica através de Computação Avançada (SciDAC) apoiado pelo DOE Office of Science com foco no avanço de software e algoritmos para reações fotoquímicas. Os projetos SciDAC normalmente reúnem uma equipe interdisciplinar de pesquisadores para desenvolver novos e inovadores métodos computacionais para lidar com alguns dos problemas científicos mais desafiadores.
"A natureza interdisciplinar da SciDAC é uma maneira muito eficaz de facilitar a descoberta da ciência, à medida que cada membro da equipe traz uma perspectiva diferente para a resolução de problemas, "diz Yang." Neste ambiente dinâmico, matemáticos, como eu, aliar-se a cientistas de domínio para identificar gargalos computacionais, em seguida, usamos técnicas matemáticas de ponta para enfrentar e superar esses desafios. "