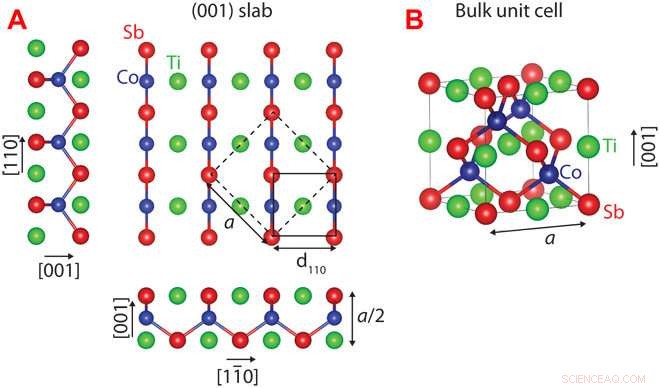
Estrutura cristalina do CoTiSb. (A) Placa não relaxada (001) de CoTiSb com terminação de TiSb. A célula unitária de massa convencional é marcada por linhas tracejadas (comprimento da borda a), e a célula unitária de superfície (1 × 1) é marcada por uma linha contínua. (B) Célula unitária cúbica convencional que consiste em uma sub-rede de zincblende CoSb preenchida com Ti. Crédito: Avanços da Ciência (2018). DOI:10.1126 / sciadv.aar5832
Quem diria que desvendar o mistério de como átomos infinitesimalmente pequenos se organizam nas bordas dos cristais em materiais avançados poderia ser tão simples quanto um, dois, três?
A modelagem da estrutura molecular da superfície de um cristal normalmente requer computadores poderosos, mas os engenheiros da Universidade de Wisconsin-Madison desenvolveram um método muito mais simples - tão fácil quanto contar com lápis e papel.
A estratégia simples pode ajudar a criar chips de computador ultrarrápidos baseados em materiais diferentes do silício.
"Ficamos surpresos ao descobrir que era, na verdade, tão simples, "diz Jason Kawasaki, um professor de ciência e engenharia de materiais da UW – Madison. "Com alguns pequenos ajustes, podíamos prever estruturas que eram quantitativamente muito precisas. "
Eles eram tão precisos que sua nova abordagem de previsão, publicado em 1º de junho, 2018 no jornal Avanços da Ciência , oferece um procedimento rápido e fácil para enfocar materiais promissores para uso em eletrônica avançada, como computadores quânticos que resolvem problemas muito mais rapidamente do que as máquinas convencionais à base de silício.
"Antes de poder usar materiais de maneiras interessantes para dispositivos de última geração, você precisa entender como a estrutura muda na superfície, "diz Kawasaki.
A previsão precisa das estruturas da superfície do cristal é um problema que há muito atormenta os cientistas. Os átomos na borda de um material tendem a se reorganizar, às vezes perdendo suas propriedades eletrônicas ou magnéticas.
Kawasaki e seus colegas se concentraram em um tipo de material chamado compostos de meio-Heusler, que têm várias propriedades eletrônicas e magnéticas sintonizáveis. Infelizmente, muitos meio-Heuslers não têm o desempenho previsto quando são combinados com outros materiais ou reduzidos a uma superfície plana.
"Quando você tem pequenos rearranjos de átomos, você pode ter grandes mudanças de propriedades, "diz Kawasaki.
Todos os materiais são feitos de átomos, que têm núcleos em seus centros rodeados por nuvens em constante mutação de minúsculas partículas subatômicas chamadas elétrons. Os átomos podem se conectar, ou vínculo, compartilhando alguns de seus elétrons uns com os outros. Os cristais consistem em muitos átomos ligados entre si em um padrão repetitivo e regularmente organizado. Esse padrão quebra, Contudo, em superfícies de cristal ou interfaces, deixando alguns átomos sem parceiros e elétrons não compartilhados pendurados longe do material a granel.
Dentro dos rígidos interiores dos cristais, simulações sofisticadas podem determinar arranjos atômicos, mas os computadores precisam de melhores suposições iniciais nas configurações para criar previsões estruturais.
Por muito tempo, as melhores suposições na superfície eram impossíveis de se obter porque a presença de elétrons pendurados faz com que o número de conformações possíveis suba rapidamente.
"As ferramentas certas e a estrutura teórica certa não existiam, "diz Kawasaki.
A estrutura teórica certa acabou sendo surpreendentemente simples, regido pelas regras básicas da química. Tudo o que é necessário é contar todos os elétrons que cada átomo traz para a superfície, calcule todos os elétrons previstos para estarem em ligações, e determinar se esses números correspondem. Quando todos os elétrons são contabilizados, a estrutura provavelmente será estável. Se não, está de volta à prancheta.
A contagem é tão direta que Kawasaki pode literalmente usar lápis e papel para fazer os cálculos.
As regras de contagem funcionam bem para materiais simples. Contudo, os cientistas presumiram que as nuvens de elétrons para os átomos metálicos que compõem os materiais meio-Heusler eram complicadas demais para essa contabilidade básica.
Kawasaki e seus colegas provaram que essa noção estava errada.
"Descobrimos que muitas das regras gerais que foram desenvolvidas para compreender a ligação em sistemas simples podem ser mapeadas nesses materiais mais complexos, "diz Kawasaki.
Usando essa abordagem, Kawasaki e colegas previram e confirmaram a configuração da superfície de um importante material de meio-Heusler chamado antimônio cobalto-titânio, que é um semicondutor potencialmente útil. Os pesquisadores mediram a superfície do cristal com técnicas avançadas de imagem, observando suas previsões em papel e lápis alinhadas perfeitamente com as configurações atômicas reais.
Os pesquisadores então aplicaram seu método a mais dois compostos de meio-Heusler, um semimetal e um ferromagneto, e planejam identificar materiais mais promissores.
Kawasaki realizou experimentos de crescimento e medição de cristal em colaboração com Chris Palmstrøm, um membro do corpo docente em engenharia elétrica e da computação e ciência dos materiais na Universidade da Califórnia, Santa Barbara.