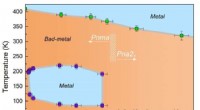
A distribuição eletrônica tetraédrica de uma molécula de água. O núcleo do átomo de oxigênio está no centro do tetraedro, e os núcleos de hidrogênio estão no centro das esferas rosa. Fundação Simons. Crédito:Simons Foundation
Uma nova ferramenta de aprendizado de máquina pode calcular a energia necessária para fazer - ou quebrar - uma molécula com maior precisão do que os métodos convencionais. Embora a ferramenta atualmente só possa lidar com moléculas simples, ele abre o caminho para futuros insights em química quântica.
"Usar o aprendizado de máquina para resolver as equações fundamentais que regem a química quântica é um problema aberto há vários anos, e há muito entusiasmo em torno disso agora, "diz o co-criador Giuseppe Carleo, um cientista pesquisador do Centro de Física Quântica Computacional do Flatiron Institute na cidade de Nova York. Uma melhor compreensão da formação e destruição de moléculas, ele diz, poderia revelar o funcionamento interno das reações químicas vitais para a vida.
Carleo e colaboradores Kenny Choo da Universidade de Zurique e Antonio Mezzacapo do IBM Thomas J. Watson Research Center em Yorktown Heights, Nova york, apresentar seu trabalho 12 de maio em Nature Communications .
A ferramenta da equipe estima a quantidade de energia necessária para montar ou separar uma molécula, como água ou amônia. Esse cálculo requer a determinação da estrutura eletrônica da molécula, que consiste no comportamento coletivo dos elétrons que unem a molécula.
A estrutura eletrônica de uma molécula é uma coisa complicada de calcular, exigindo a determinação de todos os estados potenciais em que os elétrons da molécula poderiam estar, mais a probabilidade de cada estado.
Uma vez que os elétrons interagem e tornam-se mecanicamente emaranhados uns com os outros, os cientistas não podem tratá-los individualmente. Com mais elétrons, mais complicações surgem, e o problema fica exponencialmente mais difícil. Não existem soluções exatas para moléculas mais complexas do que os dois elétrons encontrados em um par de átomos de hidrogênio. Mesmo as aproximações lutam com a precisão quando envolvem mais do que alguns elétrons.
Um dos desafios é que a estrutura eletrônica de uma molécula inclui estados para um número infinito de orbitais indo cada vez mais longe dos átomos. Adicionalmente, um elétron é indistinguível de outro, e dois elétrons não podem ocupar o mesmo estado. A última regra é uma consequência da simetria de troca, que governa o que acontece quando partículas idênticas mudam de estado.
Mezzacapo e colegas da IBM Quantum desenvolveram um método para restringir o número de orbitais considerados e impor simetria de troca. Esta abordagem, com base em métodos desenvolvidos para aplicativos de computação quântica, torna o problema mais parecido com cenários onde os elétrons estão confinados a locais predefinidos, como em uma estrutura rígida.
A semelhança com redes rígidas foi a chave para tornar o problema mais gerenciável. Carleo treinou redes neurais anteriormente para reconstruir o comportamento dos elétrons confinados aos locais de uma rede. Ao estender esses métodos, os pesquisadores puderam estimar soluções para os problemas compactados de Mezzacapo. A rede neural da equipe calcula a probabilidade de cada estado. Usando esta probabilidade, os pesquisadores podem estimar a energia de um determinado estado. O menor nível de energia, apelidado de energia de equilíbrio, é onde a molécula é mais estável.
As inovações da equipe tornaram o cálculo da estrutura eletrônica de uma molécula básica mais simples e rápido. Os pesquisadores demonstraram a precisão de seus métodos estimando quanta energia seria necessária para separar uma molécula do mundo real, quebrando suas amarras. Eles executaram cálculos para dihidrogênio (H 2 ), hidreto de lítio (LiH), amônia (NH 3 ), água (H 2 O), carbono diatômico (C 2 ) e dinitrogênio (N 2 ) Para todas as moléculas, as estimativas da equipe provaram ser altamente precisas, mesmo em intervalos onde os métodos existentes são difíceis.
No futuro, os pesquisadores pretendem lidar com moléculas maiores e mais complexas usando redes neurais mais sofisticadas. Um dos objetivos é lidar com produtos químicos como os encontrados no ciclo do nitrogênio, em que processos biológicos constroem e quebram moléculas baseadas em nitrogênio para torná-las utilizáveis para a vida. “Queremos que esta seja uma ferramenta que possa ser usada por químicos para processar esses problemas, "Carleo diz.
Carleo, Choo e Mezzacapo não estão sozinhos no aprendizado de máquina para resolver problemas em química quântica. Os pesquisadores apresentaram seu trabalho no arXiv.org pela primeira vez em setembro de 2019. No mesmo mês, um grupo na Alemanha e outro na DeepMind do Google em Londres, cada um divulgou pesquisas usando aprendizado de máquina para reconstruir a estrutura eletrônica das moléculas.
Os outros dois grupos usam uma abordagem semelhante que não limita o número de orbitais considerados. Essa inclusão, Contudo, é mais exigente computacionalmente, uma desvantagem que só vai piorar com moléculas mais complexas. Com os mesmos recursos computacionais, a abordagem de Carleo, Choo e Mezzacapo produzem maior precisão, mas as simplificações feitas para obter essa precisão podem introduzir vieses.
"Geral, é uma troca entre preconceito e precisão, e não está claro qual das duas abordagens tem mais potencial para o futuro, "Carleo diz." Só o tempo nos dirá quais dessas abordagens podem ser escaladas para os desafiadores problemas abertos na química. "