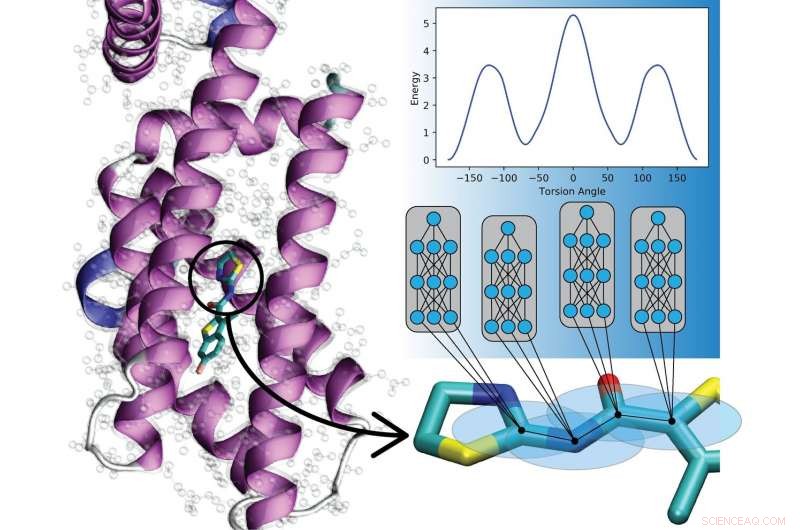
Novos modelos de aprendizado profundo prevêem as interações entre átomos em moléculas orgânicas. Esses modelos ajudarão biólogos computacionais e pesquisadores de desenvolvimento de drogas a compreender e tratar doenças. Crédito:Laboratório Nacional de Los Alamos
Novo trabalho do Laboratório Nacional de Los Alamos, a Universidade da Carolina do Norte em Chapel Hill, e a Universidade da Flórida está mostrando que as redes neurais artificiais podem ser treinadas para codificar as leis da mecânica quântica para descrever os movimentos das moléculas, supercharging simulações potencialmente em uma ampla gama de campos.
"Isso significa que agora podemos modelar materiais e dinâmica molecular bilhões de vezes mais rápido em comparação com os métodos quânticos convencionais, mantendo o mesmo nível de precisão, "disse Justin Smith, Físico de Los Alamos e Bolsista Metropolis na Divisão Teórica do Laboratório. Compreender como as moléculas se movem é fundamental para explorar seu valor potencial para o desenvolvimento de drogas, simulações de proteínas e química reativa, por exemplo, e tanto a mecânica quântica quanto os métodos experimentais (empíricos) alimentam as simulações.
A nova técnica, chamado de potencial ANI-1ccx, promete avançar as capacidades dos pesquisadores em muitos campos e melhorar a precisão dos potenciais baseados em aprendizado de máquina em estudos futuros de ligas metálicas e física de detonação.
Algoritmos de mecânica quântica (QM), usado em computadores clássicos, pode descrever com precisão os movimentos mecânicos de um composto em seu ambiente operacional. Mas QM escala muito mal com tamanhos moleculares variados, limitando severamente o escopo das simulações possíveis. Mesmo um ligeiro aumento no tamanho molecular dentro de uma simulação pode aumentar drasticamente a carga computacional. Assim, os profissionais costumam recorrer ao uso de informações empíricas, que descreve o movimento dos átomos em termos da física clássica e das Leis de Newton, permitindo simulações que escalam para bilhões de átomos ou milhões de compostos químicos.
Tradicionalmente, os potenciais empíricos tiveram de encontrar uma compensação entre precisão e transferibilidade. Quando os muitos parâmetros do potencial são ajustados para um composto, a precisão diminui em outros compostos.
Em vez de, a equipe de Los Alamos, com a University of North Carolina em Chapel Hill e a University of Florida, desenvolveu uma abordagem de aprendizado de máquina chamada aprendizado de transferência que os permite construir potenciais empíricos aprendendo a partir de dados coletados sobre milhões de outros compostos. A nova abordagem com o potencial empírico de aprendizado de máquina pode ser aplicada a novas moléculas em milissegundos, permitindo a pesquisa de um número muito maior de compostos em escalas de tempo muito mais longas.