Um autoencoder variacional profundo para análise de dados de espectrometria de massa proteômica
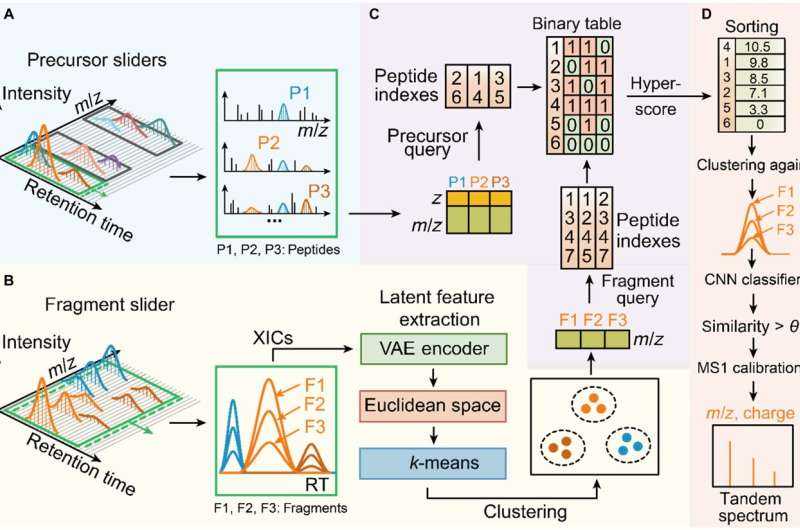
Diagrama esquemático do Dear-DIA. Crédito:Pesquisa A equipe de Jianwei Shuai e a equipe de Jiahuai Han na Universidade de Xiamen desenvolveram um software de análise de dados de aquisição independente de dados baseado em autoencoder profundo para espectrometria de massa de proteínas, que realiza a análise de peptídeos e proteínas relevantes a partir de dados complexos de espectrometria de massa de proteínas e demonstra a superioridade e versatilidade do método em diferentes instrumentos e amostras de espécies. O estudo foi publicado em Research como "Caro-DIA
XMBD
:autoencoder profundo para proteômica de aquisição independente de dados".
As proteínas desempenham um papel fundamental como executoras das atividades vitais celulares, conduzindo uma infinidade de processos biológicos cruciais. Consequentemente, o campo da proteômica tem recebido ampla atenção. A proteômica envolve o estudo abrangente das propriedades das proteínas, incluindo modificações pós-traducionais, níveis de expressão proteica, interações proteína-proteína e muito mais. Seu objetivo geral é obter uma compreensão holística da patogênese da doença, do metabolismo celular e de outros processos vitais no nível das proteínas.
Entre as principais técnicas analíticas na pesquisa proteômica, a espectrometria de massa de proteínas se destaca como a mais crítica. Com o tempo, a tecnologia de espectrometria de massa evoluiu para fornecer aos pesquisadores ferramentas confiáveis e dinâmicas para análise proteômica.
Duas abordagens principais para espectrometria de massa de proteínas são aquisição dependente de dados (DDA) e aquisição independente de dados (DIA). No DDA, todos os espectros de íons precursores de peptídeos (MS1) são adquiridos no modo de varredura completa, seguido pela seleção dos íons peptídicos mais intensivos em N para fragmentação para obter espectros de íons de fragmentos (MS2).
Apesar de sua utilidade, o DDA enfrenta desafios relacionados à reprodutibilidade experimental e detecção de peptídeos de baixa abundância devido à aleatoriedade da fragmentação peptídica e à seleção preferencial de peptídeos de alta intensidade.
Para superar estas limitações, o método de aquisição DIA foi introduzido. Esta técnica divide a faixa de razão massa-carga dos espectros de íons pais em múltiplas janelas e fragmenta sequencialmente todos os peptídeos dentro de cada janela para obter espectros de íons filhos. Um método DIA comum é a aquisição de janela sequencial de todos os íons de fragmentos teóricos (SWATH). Comparação dos resultados da análise no conjunto de dados SGS humano e no conjunto de dados de células L929 de camundongo. Crédito:Pesquisa Embora os dados de aquisição do DIA retenham informações proteômicas mais abrangentes, seu grande tamanho de dados, alta dimensionalidade e sinais espectrais complexos representam desafios para sua análise. Como resultado, a mineração de dados DIA tornou-se um foco importante na comunidade proteômica.
A equipe de Jianwei Shuai e a equipe de Jiahuai Han colaboraram para desenvolver o Dear-DIA, um software de análise de dados de aquisição independente de dados baseado em aprendizado profundo, que realiza a identificação de íons fragmentos correspondentes a diferentes peptídeos de espectros complexos de aquisição de DIA e demonstra a generalização para amostras complexas de diferentes espécies.
Dear-DIA primeiro divide os espectros em um controle deslizante de largura fixa com uma largura fixa ao longo da direção do tempo de retenção (RT), e cada controle deslizante contém um conjunto de espectros precursores MS1 e espectros de fragmentos MS2 como unidade mínima de processamento. Em seguida, um algoritmo de localização de pico foi usado para remover os íons de fundo de baixo sinal-ruído e reter os íons precursores candidatos e os íons fragmentos candidatos.
Em seguida, Dear-DIA usa um autoencoder variacional para extrair as características de pico dos íons fragmentados e mapeia as características no espaço euclidiano e, em seguida, agrupa as características, com diferentes classes de fragmentos correspondendo a diferentes peptídeos, realizando assim o processo de deconvolução do espectrograma.
Dear-DIA inclui um algoritmo de indexação chamado PIndex, que combina os precursores com os resultados do agrupamento de fragmentos e seleciona os melhores resultados de emparelhamento por meio de pontuação. Dear-DIA usa uma rede neural convolucional para recalcular a similaridade do formato do pico de fragmentos da mesma classe para eliminar íons interferentes e agrupar resultados com baixa similaridade.
Os autores testaram primeiro o desempenho do Dear-DIA em um conjunto de dados SGS Human contendo 422 peptídeos sintéticos de padrões marcados com isótopos estáveis divididos em 10 gradientes de diluição (de diluição de 1 a 512 vezes), e os dados do DIA foram obtidos em um AB Espectrômetro de massa SCIEX TTOF5600 usando a técnica SWATH para obter dados DIA. Comparação das distribuições do número de peptídeos resultantes da análise do conjunto de dados HYE124 TTOF6600 64var. Crédito:Pesquisa Os resultados da análise mostraram que o Dear-DIA encontrou mais peptídeos sintéticos em todas as soluções diluídas em comparação com os dois métodos analíticos comumente usados, Spectronaut 14 e DIA-Umpire. Os autores também compararam o número de peptídeos e proteínas encontrados pelos diferentes métodos analíticos para os conjuntos de dados SGS Human e L929 Mouse. Os resultados mostraram que o Dear-DIA foi capaz de encontrar mais peptídeos e proteínas em comparação com o Spectronaut 14 e o DIA-Umpire, cobrindo mais de 85% dos seus resultados.
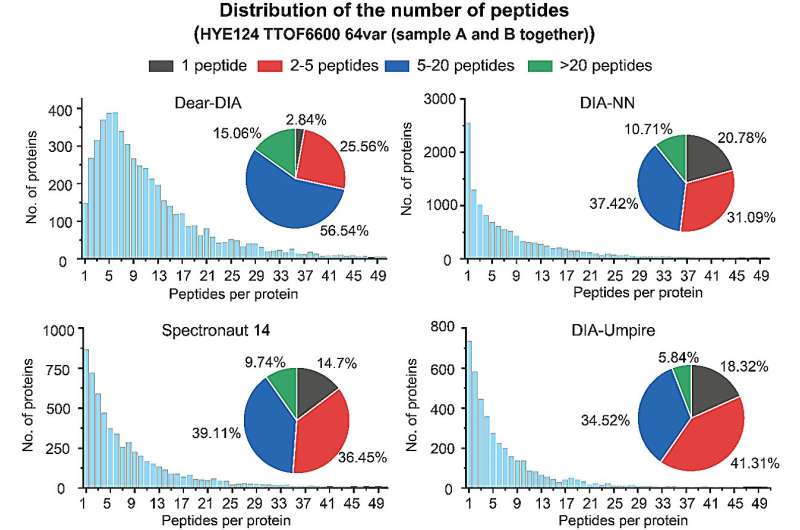
A confiança dos resultados da análise proteômica também pode ser demonstrada pelo número de peptídeos identificados para cada proteína. Proteínas com 2 ou mais peptídeos identificados são geralmente consideradas identificações mais confiáveis. Os autores compararam o número de proteínas versus peptídeos relatados por Dear-DIA com software existente em um conjunto de dados de espécies mistas (conjunto de dados HYE124 TTOF6600 64var).
O conjunto de dados contém proteínas de três espécies, humana, levedura e E. coli, e os dados foram adquiridos em um espectrômetro de massa AB SCIEX TTOF6600 usando o método SWATH, com espectros de íons parentais contendo 64 janelas variáveis. Os resultados da análise mostraram que 97,16% das proteínas encontradas por Dear-DIA poderiam corresponder a 2 ou mais peptídeos, o que é muito superior ao DIA-NN, Spectronaut 14 e DIA-Umpire.
Técnicas de aquisição independentes de dados para proteômica foram amplamente adotadas, e algoritmos de análise relacionados tornaram-se um ponto importante de pesquisa. A descoberta de proteínas a partir de dados massivos de espectrometria de massa é uma tarefa interessante e desafiadora. Neste artigo, a equipe desenvolveu o Dear-DIA, um software de análise baseado em aprendizado profundo, que é usado para processar uma variedade de dados de aquisição de DIA altamente complexos, e pode descobrir mais peptídeos e proteínas, além de reproduzir a maioria dos resultados de Espectronauta e DIA-Árbitro.
Além disso, embora o conjunto de dados de treinamento seja de E. coli, o excelente desempenho do Dear-DIA no conjunto de dados de espécies mistas demonstra sua forte capacidade de generalização para analisar dados proteômicos complexos. A aprendizagem profunda, como ferramenta amplamente utilizada para análise de big data, demonstrou excelentes capacidades de mineração de dados para descobrir associações intrínsecas profundas em big data.
O uso de aprendizagem profunda para analisar dados de espectrometria de massa proteômica tem grande potencial e promoverá ainda mais o estudo de questões fundamentais, como redes de sinalização de proteínas.
Mais informações: Qingzu He e outros, Dear-DIA
XMBD
:Deep Autoencoder permite a desconvolução de proteômica de aquisição independente de dados, Pesquisa (2023). DOI:10.34133/pesquisa.0179 Informações do diário: Pesquisa