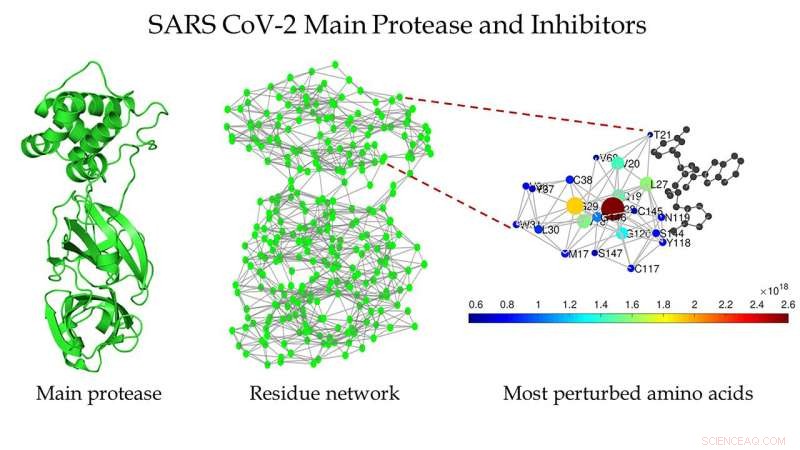
Esquema da protease principal do SARS CoV-2 (à esquerda), a rede de resíduos de proteínas da principal protease do SARS CoV-2 (centro), e uma visão ampliada da região ao redor do local de ligação, conforme detectado pela Estrada (direita). Crédito:Ernesto Estrada
Como a pandemia de COVID-19 causada pelo coronavírus SARS-CoV-2 continua a se espalhar pelo mundo, muitos pesquisadores estão estudando modelos epidemiológicos para prever sua propagação.
Contudo, Ernesto Estrada, um matemático e especialista em sistemas complexos da Fundação ARAID da Universidade de Zaragoza, decidiu se concentrar em encontrar alvos dentro do SARS-CoV-2 para novos medicamentos atacarem. Do trabalho anterior, ele sabia que a principal protease do vírus, uma enzima responsável pelo processamento proteolítico de poliproteínas, é um excelente alvo.
No jornal Caos , Estrada disse quando ele e seus colegas descobriram um aumento dramático na sensibilidade da principal protease do SARS-CoV-2 a pequenos distúrbios, isso os fez suspeitar que os inibidores desempenham um papel na eliminação do vírus.
Os inibidores são moléculas orgânicas, drogas, ou novos compostos químicos que se ligam ao local de ligação de uma protease para inibir seu trabalho. Um vírus morre sem uma enzima proteolítica trabalhando para ele.
“Percebi que os químicos já haviam encontrado alguns inibidores potentes da principal protease da SARS-CoV-2, e que eles resolveram a estrutura desta proteína por meio de cristalografia de raios-X, "disse ele." Foi chocante ver que esta protease é muito semelhante à do coronavírus SARS, que produziu a epidemia de 2003, SARS-CoV-1. "
Quando os pesquisadores sobrepuseram as duas estruturas uma sobre a outra, eles combinaram quase perfeitamente.
"Se você alinhar as sequências de aminoácidos de ambas as proteases, existem apenas 12 dos 306 resíduos que não coincidem, "Estrada disse." Existe algo escondido por trás dessas semelhanças aparentes entre as duas proteases? Podemos aprender algo com eles para melhorar o design de drogas contra o vírus? "
O grupo de Estrada tem vasta experiência na análise de redes, como redes sociais, a Internet, ou cadeias alimentares entre espécies dentro de um ambiente - e decidiu tratar uma proteína como uma rede.
"Eles são chamados de redes de resíduos de proteínas, onde representamos cada aminoácido como um nó, e a interação entre dois aminoácidos é representada por uma ligação entre os dois, " ele explicou.
Eles encontraram várias estruturas da protease principal do SARS CoV-1 e SARS CoV-2 que estavam limpas, o que significa que eles não contêm mutações, ligantes, ou solventes dentro de suas estruturas. Eles transformaram sua estrutura em redes de resíduos de proteínas.
Estrada disse que as medidas de rede mais tradicionais revelaram que as duas estruturas eram muito semelhantes entre si, algo que sua equipe já sabia. "Mas alguns anos atrás, desenvolvemos uma medida matemática mais sofisticada que nos permite detectar a que distância uma perturbação dentro de uma rede pode ser propagada. Esse trabalho era muito teórico, natureza matemática, mas especulamos que poderia ser útil para o estudo de proteínas. "
Então eles o testaram. Ele revelou que a protease de SARS-CoV-2 é 1, 900% mais sensível à transmissão de longo alcance de perturbações do que a protease de SARS-CoV-1.
"Isso significa que quando uma proteína é perturbada, por exemplo, pela água dentro do ambiente intracelular, tais perturbações são transmitidas através de uma rede de intraresídeos que formam a estrutura 3-D da proteína, "Estrada disse." Se tal perturbação for produzida em torno de um determinado aminoácido dentro da protease de SARS-CoV-1, é transmitido apenas através de um ambiente próximo ao aminoácido perturbado. "
Mas se essa perturbação ocorrer em um aminoácido dentro da protease de SARS-CoV-2, é transmitido para quase toda a rede - até mesmo para aminoácidos muito distantes.
"É notável, porque significa que, com pequenas diferenças estruturais, a protease do SARS-CoV-2 é muito mais eficaz nas comunicações intraresiduais, "Estrada disse." Deve ser muito mais eficaz em fazer seu trabalho como uma enzima proteolítica do vírus. O diabo fez um trabalho quase perfeito aqui, mas ele deixou a porta aberta. Essa grande sensibilidade da protease SARS-CoV-2 às perturbações pode ser seu calcanhar de Aquiles em relação aos inibidores. "
A abordagem do grupo pode ser usada para protocolos de triagem massiva para identificar inibidores potentes da protease principal da SARS-CoV-2 e, consequentemente, para o desenvolvimento de novos medicamentos para matá-lo.