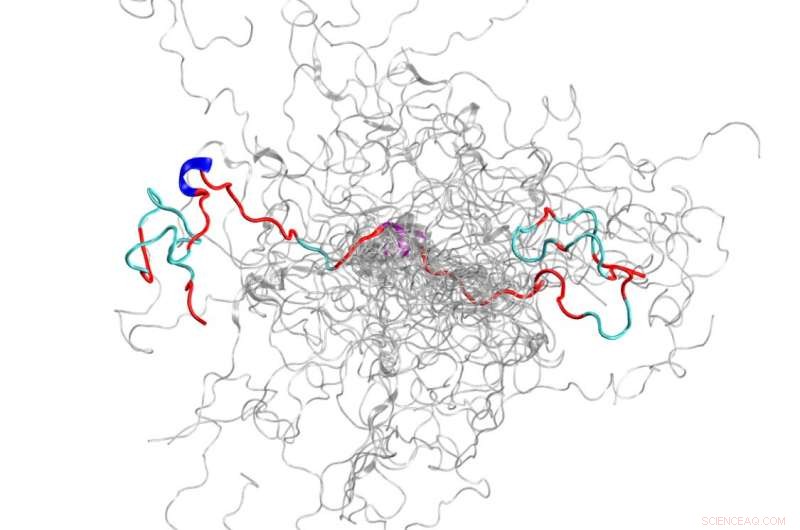
O conjunto configuracional (uma coleção de estruturas 3D) de uma proteína intrinsecamente desordenada, o N-terminal da c-Src quinase, que é a principal proteína de sinalização em humanos. Crédito:Oak Ridge National Laboratory, Departamento de Energia dos EUA
Usando o supercomputador Titan e a Spallation Neutron Source no Oak Ridge National Laboratory do Departamento de Energia, os cientistas criaram o modelo 3-D mais preciso de uma proteína intrinsecamente desordenada, revelando o conjunto de suas estruturas de nível atômico.
Como seu nome indica, um IDP não adota um ordenado, estrutura estática como outras proteínas; em vez de, é flexível e pode adotar várias estruturas 3-D. Essa falta de uma estrutura única é necessária para a função biológica do IDP, mas torna seu estudo tecnicamente desafiador. Os IDPs podem ser uma proteína inteira ou um domínio de uma proteína estruturada de outra forma, e eles constituem uma grande parte do ser humano, micróbio, e proteínas vegetais.
Loukas Petridis, um cientista da equipe do Centro de Biofísica Molecular do ORNL, direcionou uma equipe de pesquisadores para uma nova maneira de criar modelos físicos precisos de tais biossistemas flexíveis, o que pode levar a uma melhor compreensão de suas funções biológicas. Nos últimos três anos, a equipe combinou experimentos de espalhamento de nêutrons com simulações de dinâmica molecular (MD) de amostragem aprimorada tão exigentes computacionalmente que exigiram o poder de processamento de Titã, o recém-desativado Cray XK7 de 27 petaflop no Oak Ridge Leadership Computing Facility, um DOE Office of Science User Facility em ORNL.
“Estudar esses deslocados internos é bastante difícil, de ambas as perspectivas de experimentos e modelagem, "disse Utsab Shrestha, o autor principal do artigo da equipe, publicado recentemente no Proceedings of the National Academy of Sciences . "Não pensamos nisso apenas a partir de experimentos ou simulações, planejamos de forma a sinergizar ambas as abordagens - combiná-las de forma que pudéssemos obter informações mais precisas sobre os deslocados internos. Especificamente, simulações nos ajudaram a gerar um conjunto preciso de IDP em resolução atômica, o que é difícil de determinar apenas com base em experimentos. "
Tipicamente, pesquisadores realizam experimentos como espalhamento de nêutrons de pequeno ângulo, espalhamento de raios-X de baixo ângulo, ou ressonância magnética nuclear para sondar sistemas biológicos flexíveis. Contudo, esses métodos não fornecem uma imagem detalhada de nível atômico das estruturas 3-D de um IDP, conhecido como seu conjunto configuracional. Além disso, eles só podem produzir dados com média de conjunto, em vez das configurações específicas da estrutura da proteína subjacente. Os cientistas também realizaram simulações de computador de IDP e as compararam com tais experimentos, na esperança de obter os mesmos resultados para verificar a precisão de seus modelos.
“Mas eles acabam não concordando com os experimentos, "Petridis disse." E por causa da discrepância entre as simulações e os experimentos, eles têm que pesar novamente as simulações - eles têm que ajustar os resultados da simulação para fazê-los corresponder aos experimentos, o que é frustrante. Esse era o estado da arte até o nosso trabalho. "
As simulações computadorizadas de MD conduzidas por Shrestha usaram métodos de amostragem aprimorados que tiveram sucesso em combinar não apenas experimentos de espalhamento de nêutrons - conduzidos por Viswanathan Gurumoorthy e seus colegas no SNS, um DOE Office of Science User Facility no ORNL - mas também dados de NMR publicados anteriormente. Essas simulações de MD usam física para determinar como as proteínas se movem. A chave para o sucesso da equipe foi a execução de muitas simulações de MD em paralelo no Titan, permitindo que as simulações se comuniquem entre si e troquem informações.
"Isso é muito importante porque permite que a simulação faça uma amostra de um espaço configuracional maior, explorar mais as estruturas tridimensionais de uma forma mais eficiente, "Petridis disse." É por isso que este MD de amostragem aprimorada pode produzir resultados que a simulação de MD normal não pode. Teríamos que executar uma simulação normal de MD durante anos para obter os mesmos resultados. "
O IDP que a equipe escolheu para estudar é o domínio N-terminal da c-Src quinase, que é a principal proteína de sinalização em humanos. Mutações nesta proteína complexa foram correlacionadas com câncer, o que também o torna um importante alvo de drogas. Ao mapear este domínio anteriormente obscuro, os cientistas foram capazes de descobrir novas informações sobre suas estruturas 3-D que métodos anteriores não haviam mostrado. Por exemplo, embora seja bastante desordenado, esta proteína forma estruturas ordenadas transitórias, como hélices.
"A combinação de experimentos de espalhamento de nêutrons e simulação é muito poderosa, "Disse Petridis." A validação das simulações por comparação com experimentos de espalhamento de nêutrons é essencial para ter confiança nos resultados da simulação. As simulações validadas podem fornecer informações detalhadas que não são obtidas diretamente por meio de experimentos. "
O modelo de computador detalhado do conjunto de estrutura 3-D do IDP abre a porta para mais experimentação. Por exemplo, os cientistas poderiam simular o efeito da fosforilação (a adição de um grupo fosfato à proteína que pode regular a função da proteína) para ver quais mudanças estruturais ocorrem na c-Src quinase que podem influenciar sua função. O papel das mutações também pode ser examinado:se um pesquisador alterar um aminoácido da cadeia, como isso afeta a estrutura ou o conjunto de estruturas?
"Há muitas perguntas não respondidas para c-Src quinase em particular que poderiam ser respondidas em termos de interações com outros parceiros - o efeito da fosforilação, o efeito das mutações, "Petridis disse.
Além dos potenciais usos científicos para o próprio modelo, Petridis vê oportunidades de aplicar o uso de computação de alto desempenho para executar MD de amostragem aprimorada para estudar as estruturas de muitos outros IDPs importantes, o que pode dar uma visão sobre sua função. E de forma mais ampla, a equipe quer desenvolver tecnologias de simulação que podem reproduzir perfis de espalhamento de nêutrons de pequeno ângulo de sistemas biológicos ainda mais complexos.
"Não queremos investigar apenas as proteínas desordenadas - queremos sistemas muito maiores que contenham domínios ordenados e desordenados que podem interagir com membranas ou DNA, "Petridis disse." A dispersão de nêutrons é, na minha opinião, a melhor técnica experimental para sondar esses sistemas de múltiplos componentes - por exemplo, uma proteína que interage com uma membrana ou uma proteína que interage com o DNA. Mas, ainda, o espalhamento de nêutrons precisa de simulações precisas para interpretar melhor os dados. "