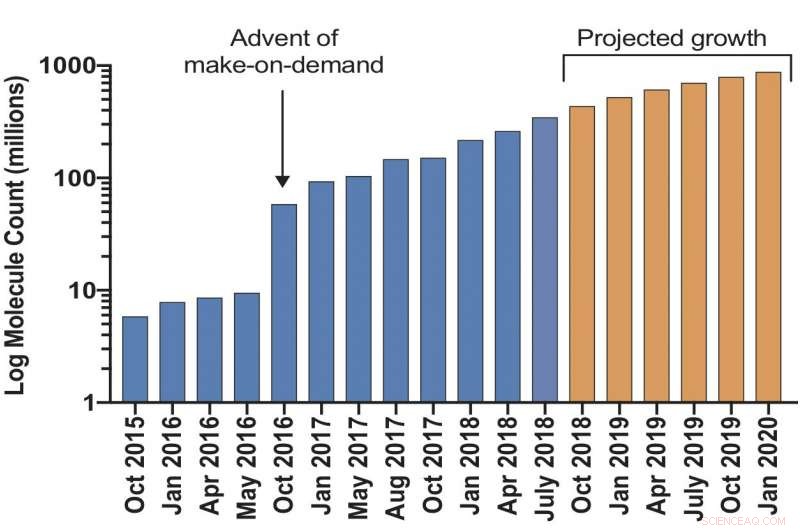
Espera-se que uma biblioteca virtual de moléculas sob demanda disponíveis para a descoberta de medicamentos ultrapasse 1 bilhão de compostos no próximo ano. Crédito:Bryan Roth, M.D., Ph.D., da Universidade da Carolina do Norte (UNC) Chapel Hill, Brian Shoichet, Ph.D., e John Irwin, Ph.D., da University of California San Francisco, e colegas.
Os pesquisadores lançaram uma biblioteca de dock virtual ultragrande que deve crescer para mais de 1 bilhão de moléculas no próximo ano. Ele irá expandir em 1000 vezes o número de tais compostos "feitos sob demanda" prontamente disponíveis para cientistas para biologia química e descoberta de drogas. Quanto maior a biblioteca, maiores serão suas chances de eliminar moléculas "iscas" inativas que poderiam levar os pesquisadores a becos sem saída. O projeto é financiado pelo National Institutes of Health.
"Para melhorar os medicamentos para doenças mentais, precisamos rastrear um grande número de moléculas potencialmente terapêuticas, "explicou Joshua A. Gordon, M.D., Ph.D., diretor do Instituto Nacional de Saúde Mental do NIH (NIMH), que co-financiou a pesquisa. "A modelagem computacional imparcial nos permite fazer isso em um computador, acelerando enormemente o processo de descoberta de novos tratamentos. Ele permite que os pesquisadores virtualmente "vejam" uma molécula atracando com sua proteína receptora - como um navio em seu cais ou uma chave em sua fechadura - e prevejam suas propriedades farmacológicas, com base em como as estruturas moleculares devem interagir. Apenas aquelas relativamente poucas moléculas candidatas que melhor correspondem ao perfil do alvo no computador precisam ser feitas fisicamente e testadas em um laboratório úmido. "
Bryan Roth, M.D., Ph.D., da Universidade da Carolina do Norte (UNC) Chapel Hill, Brian Shoichet, Ph.D., e John Irwin, Ph.D., da University of California San Francisco, e colegas, relatório sobre suas descobertas em 6 de fevereiro, 2019 no jornal Natureza . O estudo foi apoiado, em parte, por doações do NIMH, Instituto Nacional de Ciências Médicas Gerais (NIGMS), o Fundo Comum do NIH, e Instituto Nacional de Doenças Neurológicas e Derrame (NINDS).
O programa Illuminating the Druggable Genome (IDG) do Fundo Comum do NIH - lançado em 2014 para catalisar pesquisas sobre proteínas que atualmente não são estudadas e são alvos potenciais de intervenção terapêutica - financiou a expansão da biblioteca docking.
Ao longo dos últimos anos, Roth, Shoichet, e colegas empregaram sua abordagem de acoplamento baseado em estrutura virtual para descobrir segredos moleculares de uma droga antipsicótica e LSD acoplados em seus respectivos receptores-alvo - e para criar um analgésico planejado que tem como alvo seletivo os circuitos analgésicos cerebrais sem os efeitos colaterais da morfina.
Sabe-se da existência de um número surpreendente de moléculas semelhantes a drogas em potencial. Ainda, centenas de milhões a bilhões de moléculas diversas permaneceram inacessíveis devido às limitações dos métodos existentes usados para compilar bibliotecas moleculares, dizem os pesquisadores. Por exemplo, sua técnica de encaixe baseada em estrutura virtual, embora prometendo, corre o risco de encontrar muitos falsos positivos ou "iscas - falhas no modelo permitem moléculas que parecem plausíveis, mas acabam sendo biologicamente inativas.
Seleção de moléculas descobertas usando a mega docking library. Crédito:Bryan Roth, M.D., Ph.D., da Universidade da Carolina do Norte (UNC) Chapel Hill, Brian Shoichet, Ph.D., e John Irwin, Ph.D., da University of California San Francisco, e colegas.
Para superar este desafio, os pesquisadores se concentraram em moléculas que resultam de 130 reações químicas bem caracterizadas usando 70, 000 diferentes blocos de construção químicos. Simulações de computador com essas moléculas mostraram que, à medida que o tamanho de uma biblioteca crescia, a proporção de "verdadeiros ativos" para chamarizes aumentou - assim como o poder estatístico de um estudo aumenta com uma amostra maior.
No novo estudo, os pesquisadores examinaram a ancoragem baseada na estrutura de 138 milhões de moléculas com o receptor D4, uma proteína chave que medeia as ações do mensageiro químico do cérebro dopamina, ou a enzima AmpC, que confere resistência a certos antibióticos e tem se mostrado difícil de bloquear.
"O receptor D4 é de particular interesse para o NIMH por causa de seu papel na cognição e outras funções executivas do córtex pré-frontal do cérebro que são frequentemente perturbadas em doenças mentais, "disse Laurie Nadler, Ph.D., da Divisão de Neurociência e Ciências Básicas do Comportamento do NIMH, oficial do programa para a bolsa de apoio ao estudo do receptor D4.
Os pesquisadores então sintetizaram e testaram, em um laboratório, as 549 moléculas principais que virtualmente se encaixaram melhor no receptor D4 e 44 moléculas que se encaixaram melhor na enzima. Esses estudos revelaram várias novas moléculas semelhantes a drogas que se ligam apenas ao receptor D4 (e não aos receptores de dopamina D2 ou D3 intimamente relacionados) e ligam ou desligam o receptor. Adicionalmente, uma molécula (4163) emergiu como o aglutinante mais potente de AmpC de todos os tempos. A classificação de acoplamento de uma molécula virtual previu sua probabilidade real de se ligar ao receptor de dopamina D4 em um ensaio de laboratório.
A descoberta de moléculas novas e potentes para ambos os alvos também confirmou que as bibliotecas ultra-grandes contêm moléculas mais adequadas a uma determinada estrutura de receptor do que bibliotecas menores e que o docking virtual pode reconhecer as moléculas e prever o número total de compostos ativos esperados dentro de uma biblioteca.
"Este novo estudo ilustra o potencial de triagem computacional imparcial e docking molecular para descobrir novas moléculas de ferramenta e potenciais agentes terapêuticos, fornecendo um caminho rápido e robusto que levará diretamente a novos tratamentos com drogas para doenças mentais, "acrescentou Gordon.