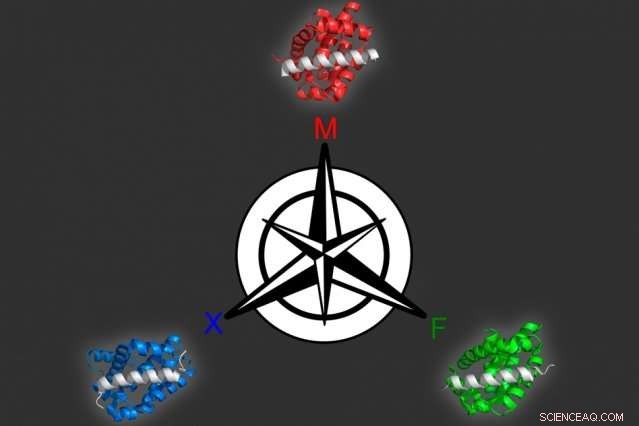
Usando uma abordagem de modelagem de computador que eles desenvolveram, Os biólogos do MIT identificaram três proteínas diferentes que podem se ligar seletivamente a cada um dos três alvos semelhantes, todos os membros da família de proteínas Bcl-2. Crédito:Vincent Xue
Projetar proteínas sintéticas que podem atuar como drogas para o câncer ou outras doenças pode ser um processo tedioso:geralmente envolve a criação de uma biblioteca de milhões de proteínas, em seguida, rastrear a biblioteca para encontrar proteínas que se ligam ao alvo correto.
Os biólogos do MIT agora criaram uma abordagem mais refinada, na qual usam modelagem de computador para prever como diferentes sequências de proteínas irão interagir com o alvo. Esta estratégia gera um maior número de candidatos e também oferece maior controle sobre uma variedade de características de proteínas, diz Amy Keating, professor de biologia e engenharia biológica e líder da equipe de pesquisa.
"Nosso método oferece um campo de jogo muito maior, onde você pode selecionar soluções que são muito diferentes umas das outras e terão diferentes pontos fortes e desvantagens, "ela diz." Nossa esperança é que possamos fornecer uma gama mais ampla de soluções possíveis para aumentar o rendimento desses acessos iniciais em algo útil, moléculas funcionais. "
Em um jornal publicado no Proceedings of the National Academy of Sciences a semana de 15 de outubro, Keating e seus colegas usaram essa abordagem para gerar vários peptídeos que podem ter como alvo diferentes membros de uma família de proteínas chamada Bcl-2, que ajudam a impulsionar o crescimento do câncer.
Destinatários recentes de PhD Justin Jenson e Vincent Xue são os principais autores do artigo. Outros autores são pós-doutorado Tirtha Mandal, ex-técnico de laboratório Lindsey Stretz, e o ex-pós-doutorado Lothar Reich.
Modelagem de interações
Drogas proteicas, também chamados de biofármacos, são uma classe de medicamentos em rápido crescimento que são promissores para o tratamento de uma ampla gama de doenças. O método usual para identificar essas drogas é examinar milhões de proteínas, escolhidos aleatoriamente ou selecionados pela criação de variantes de sequências de proteínas que já se mostraram candidatas promissoras. Isso envolve a engenharia de vírus ou leveduras para produzir cada uma das proteínas, em seguida, expô-los ao alvo para ver quais ligam melhor.
"Essa é a abordagem padrão:completamente aleatória, ou com algum conhecimento prévio, projetar uma biblioteca de proteínas, e depois pescar na biblioteca para tirar os membros mais promissores, "Keating diz.
Embora esse método funcione bem, geralmente produz proteínas que são otimizadas para apenas um único traço:quão bem ele se liga ao alvo. Não permite nenhum controle sobre outros recursos que podem ser úteis, como características que contribuem para a capacidade de uma proteína de entrar nas células ou sua tendência de provocar uma resposta imunológica.
"Não há uma maneira óbvia de fazer esse tipo de coisa - especificar um peptídeo com carga positiva, por exemplo, usando a triagem da biblioteca de força bruta, "Keating diz.
Outra característica desejável é a capacidade de identificar proteínas que se ligam fortemente ao seu alvo, mas não a alvos semelhantes, o que ajuda a garantir que os medicamentos não tenham efeitos colaterais indesejados. A abordagem padrão permite que os pesquisadores façam isso, mas os experimentos se tornam mais complicados, Keating diz.
A nova estratégia envolve primeiro a criação de um modelo de computador que pode relacionar sequências de peptídeos à sua afinidade de ligação para a proteína alvo. Para criar este modelo, os pesquisadores primeiro escolheram cerca de 10, 000 peptídeos, cada 23 aminoácidos de comprimento e estrutura helicoidal, e testou sua ligação a três membros diferentes da família Bcl-2. Eles escolheram intencionalmente algumas sequências que já sabiam que se ligariam bem, além de outros que eles sabiam que não, portanto, o modelo pode incorporar dados sobre uma variedade de capacidades de ligação.
A partir deste conjunto de dados, o modelo pode produzir uma "paisagem" de como cada sequência de peptídeo interage com cada alvo. Os pesquisadores podem então usar o modelo para prever como outras sequências irão interagir com os alvos, e gerar peptídeos que atendam aos critérios desejados.
Usando este modelo, os pesquisadores produziram 36 peptídeos que se previa que se ligariam fortemente a um membro da família, mas não aos outros dois. Todos os candidatos tiveram um desempenho extremamente bom quando os pesquisadores os testaram experimentalmente, então eles tentaram um problema mais difícil:identificar proteínas que se ligam a dois dos membros, mas não ao terceiro. Muitas dessas proteínas também tiveram sucesso.
"Esta abordagem representa uma mudança de apresentar um problema muito específico e, em seguida, projetar um experimento para resolvê-lo, investir algum trabalho inicial para gerar este cenário de como a sequência está relacionada à função, capturar a paisagem em um modelo, e, em seguida, ser capaz de explorá-lo à vontade para várias propriedades, "Keating diz.
Sagar Khare, professor associado de química e biologia química na Rutgers University, diz que a nova abordagem é impressionante em sua capacidade de discriminar alvos proteicos intimamente relacionados.
"A seletividade das drogas é crítica para minimizar os efeitos fora do alvo, e muitas vezes a seletividade é muito difícil de codificar porque existem tantos competidores moleculares de aparência semelhante que também ligam a droga à parte do alvo pretendido. Este trabalho mostra como codificar essa seletividade no próprio design, "diz Khare, que não participou da pesquisa. "Aplicações no desenvolvimento de peptídeos terapêuticos ocorrerão quase com certeza."
Drogas seletivas
Os membros da família de proteínas Bcl-2 desempenham um papel importante na regulação da morte celular programada. A desregulação dessas proteínas pode inibir a morte celular, ajudando os tumores a crescerem sem controle, muitas empresas farmacêuticas têm trabalhado no desenvolvimento de medicamentos que têm como alvo essa família de proteínas. Para que tais medicamentos sejam eficazes, pode ser importante para eles almejar apenas uma das proteínas, porque interromper todos eles pode causar efeitos colaterais prejudiciais nas células saudáveis.
"Em muitos casos, as células cancerosas parecem estar usando apenas um ou dois membros da família para promover a sobrevivência celular, "Keating diz." Em geral, reconhece-se que ter um painel de agentes seletivos seria muito melhor do que uma ferramenta tosca que simplesmente os derrubaria. "
Os pesquisadores entraram com pedido de patentes para os peptídeos que identificaram neste estudo, e eles esperam que sejam testados como possíveis drogas. O laboratório de Keating agora está trabalhando na aplicação dessa nova abordagem de modelagem a outros alvos de proteína. Este tipo de modelagem pode ser útil não apenas para o desenvolvimento de drogas potenciais, mas também gerando proteínas para uso em aplicações agrícolas ou de energia, ela diz.
Esta história foi republicada por cortesia do MIT News (web.mit.edu/newsoffice/), um site popular que cobre notícias sobre pesquisas do MIT, inovação e ensino.