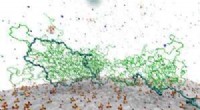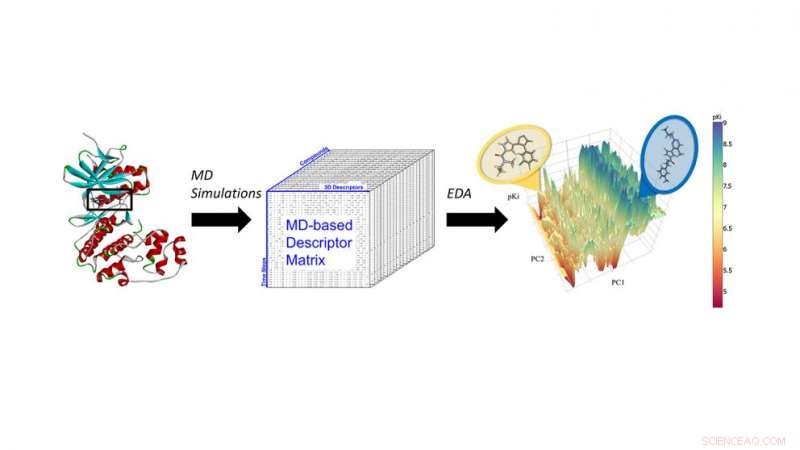
Simulações de dinâmica molecular (MD) de inibidores ERK2 para extrair descritores de MD para análise quiminformática de última geração e aprendizado de máquina. Crédito:North Carolina State University
Pesquisadores da North Carolina State University demonstraram que simulações de dinâmica molecular e técnicas de aprendizado de máquina podem ser integradas para criar modelos de previsão de computador mais precisos. Esses modelos "hiperpreditivos" podem ser usados para prever rapidamente quais novos compostos químicos podem ser candidatos a drogas promissores.
O desenvolvimento de medicamentos é um processo caro e demorado. Para reduzir o número de compostos químicos que podem ser candidatos a drogas em potencial, os cientistas utilizam modelos de computador que podem prever como um determinado composto químico pode interagir com um alvo biológico de interesse - por exemplo, uma proteína-chave que pode estar envolvida no processo de uma doença. Tradicionalmente, isso é feito por meio de modelagem quantitativa de relação estrutura-atividade (QSAR) e docking molecular, que dependem de informações 2 e 3-D sobre esses produtos químicos.
Denis Fourches, professor assistente de química computacional, queria melhorar a precisão desses modelos QSAR. “Quando você está testando um conjunto de 30 milhões de compostos, você não precisa necessariamente de uma confiabilidade muito alta com seu modelo - você está apenas tendo uma ideia aproximada sobre os 5 ou 10 por cento principais dessa biblioteca virtual. Mas se você está tentando estreitar um campo de 200 análogos para 10, que é mais comumente o caso no desenvolvimento de drogas, sua técnica de modelagem deve ser extremamente precisa. As técnicas atuais definitivamente não são confiáveis o suficiente. "
Fourches e Jeremy Ash, um estudante de graduação em bioinformática, decidiu incorporar os resultados dos cálculos de dinâmica molecular - simulações de todos os átomos de como um composto específico se move no bolso de ligação de uma proteína - em modelos de previsão baseados em aprendizado de máquina.
"A maioria dos modelos usa apenas as estruturas bidimensionais das moléculas, "Fourches diz." Mas, na realidade, produtos químicos são objetos tridimensionais complexos que se movem, vibrar e ter interações intermoleculares dinâmicas com a proteína, uma vez ancorada em seu local de ligação. Você não pode ver isso se apenas olhar para a estrutura 2-D ou 3-D de uma determinada molécula. "
Em um estudo de prova de conceito, Fourches e Ash analisaram a ERK2 quinase - uma enzima associada a vários tipos de câncer - e um grupo de 87 inibidores ERK2 conhecidos, variando de muito ativo a inativo. Eles executaram simulações de dinâmica molecular (MD) independentes para cada um desses 87 compostos e computaram informações críticas sobre a flexibilidade de cada composto uma vez no bolso ERK2. Em seguida, eles analisaram os descritores MD usando técnicas de quimio-informática e aprendizado de máquina. Os descritores de MD foram capazes de distinguir com precisão os inibidores ERK2 ativos de fracamente ativos e inativos, o que não acontecia quando os modelos usavam apenas informações estruturais 2-D e 3-D.
"Já tínhamos dados sobre essas 87 moléculas e sua atividade no ERK2, "Fourches diz." Então, testamos para ver se nosso modelo era capaz de encontrar os compostos mais ativos de forma confiável. De fato, distinguiu com precisão entre inibidores ERK2 fortes e fracos, e porque os descritores de MD codificam as interações que esses compostos criam no bolso de ERK2, também nos deu mais informações sobre por que os inibidores fortes funcionavam bem.
"Antes que os avanços da computação nos permitissem simular esse tipo de dados, levaríamos seis meses para simular uma única molécula no bolso do ERK2. Graças à aceleração da GPU, agora leva apenas três horas. Isso é uma virada de jogo. Tenho esperança de que a incorporação de dados extraídos da dinâmica molecular em modelos QSAR permitirá uma nova geração de modelos hiperpreditivos que ajudarão a trazer novidades, medicamentos eficazes no mercado ainda mais rápido. É a inteligência artificial trabalhando para que possamos descobrir as drogas de amanhã. "