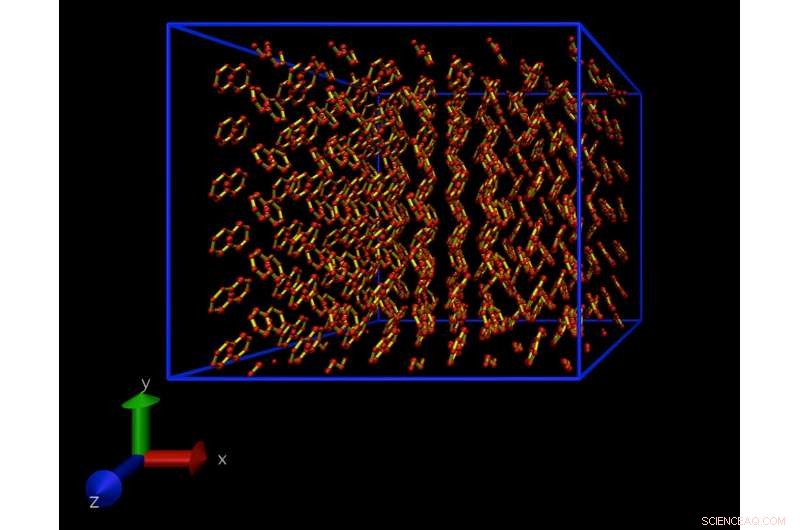
Um instantâneo de uma simulação de Dinâmica Molecular de um modelo atomístico de um cristal de naftaleno. Este cristal é repetido periodicamente em todas as direções, para eliminar os efeitos de superfície. Crédito:Daan Frenkel, Universidade de Cambridge
A solubilidade de qualquer substância - a medida de quão bem a substância se dissolve em outra substância referida como o solvente - depende de propriedades básicas como temperatura e pressão, bem como as identidades químicas da substância dissolvida (o soluto) e do solvente.
A previsão da solubilidade é importante para uma variedade de aplicações. No campo farmacêutico, por exemplo, é fundamental saber a solubilidade de um medicamento, uma vez que determina diretamente sua disponibilidade para o corpo. A indústria do petróleo fornece outro exemplo:substâncias com baixa solubilidade podem formar escamas ou depósitos indesejados em tubos ou brocas, causando bloqueios e outros grandes problemas.
Apesar da importância de prever a solubilidade, não é uma tarefa fácil. Uma abordagem, usando simulações de "força bruta", requer longos tempos de computação. Outras técnicas, enquanto mais rápido, falha em prever valores precisos de solubilidade. Esta semana em The Journal of Chemical Physics , pesquisadores relatam um novo tipo de software que permite estimativas convenientes de solubilidade de essencialmente qualquer substância molecular em amplas faixas de temperatura e pressão. O código faz uso de software de código aberto prontamente disponível e deve ser amplamente adotado.
Daan Frenkel, da Universidade de Cambridge, no Reino Unido, trabalhou com os colegas Lunna Li, também em Cambridge, e Tim Totton, da British Petroleum, para desenvolver o código.
"Fizemos uma escolha consciente de usar dados bem documentados, software disponível gratuitamente porque queríamos disponibilizar nossa abordagem para qualquer pessoa, "Frenkel disse." Uma ferramenta de propósito geral para calcular solubilidades tem estado ausente por muito tempo. A metodologia subjacente estava lá, mas ninguém havia realmente criado um programa de trabalho. "
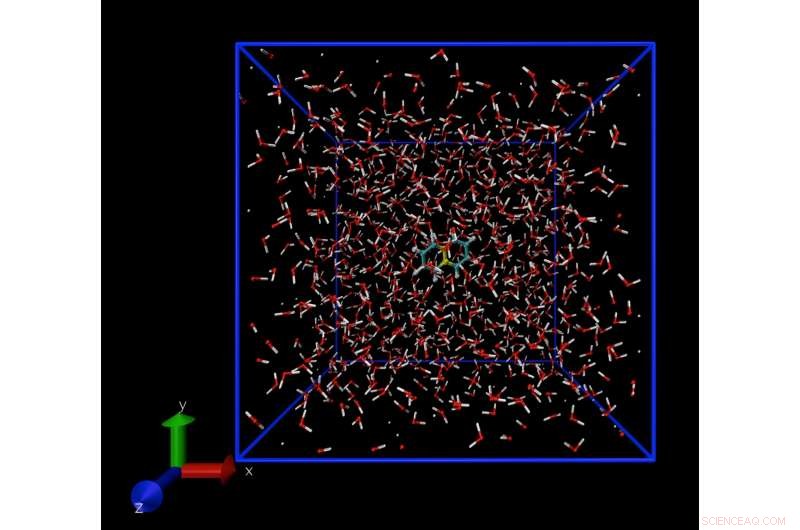
Um instantâneo de uma simulação de dinâmica molecular mostrando uma única molécula de naftaleno, dissolvido em água. A técnica de simulação permite calcular a concentração de moléculas de naftaleno em água no limite de solubilidade. Crédito:Daan Frenkel, Universidade de Cambridge
O software desenvolvido por este grupo usa expressões termodinâmicas padrão que são conhecidas desde meados do século 19, como pressão de vapor. A abordagem explora o fato de que quando uma fase sólida ou líquida está em equilíbrio, suas pressões de vapor são iguais. Quando um líquido ou sólido é aquecido, as moléculas escapam e formam vapor. Esta pressão de vapor pode ser calculada usando modelos de computador.
Por exemplo, um torrão de açúcar se dissolvendo na água:as moléculas de açúcar existem em um estado sólido - o torrão de açúcar cristalino - ou completamente rodeado por moléculas de água depois que se dissolvem. A quantidade de açúcar em cada uma das duas fases, sólido e solução, é determinado pela energia necessária para mover as moléculas de açúcar entre essas fases. A solubilidade pode ser calculada calculando a pressão de vapor das duas fases e igualando-as.
Para modelar a fase sólida, os investigadores usaram um modelo conhecido como cristal de Einstein. Neste modelo, moléculas de soluto não interagentes são colocadas em uma rede e amarradas a um ponto da rede com uma mola matemática. A pressão de vapor do cristal é calculada calculando o trabalho necessário para desligar as molas e ligar as interações entre as moléculas presas.
Para modelar uma molécula de soluto dissolvido, os investigadores usaram um potencial de energia padrão para o solvente em questão, que era água nos exemplos usados para testar o software, e calculou o trabalho em três etapas. Primeiro, uma cavidade no solvente é criada. Uma molécula de soluto é então inserida na cavidade e, finalmente, a cavidade é reduzida ao tamanho da molécula de soluto. Este procedimento elimina uma série de erros e produz estimativas precisas da pressão de vapor e, portanto, a solubilidade.
No relatório desta semana, os investigadores testaram seu código em naftaleno dissolvido em água e previram uma solubilidade que se compara bem aos valores experimentais. As investigações futuras se concentrarão em estender o software para que ele possa lidar com moléculas de soluto maiores.